

Yolanda Viana
Hiperpigmentación de las falanges distales del lactante
La hiperpigmentación de las falanges distales del recién nacido es una dermatosis benigna transitoria de la infancia, autorresolutiva y no relacionada con otros trastornos cutáneos. Es un hallazgo habitual en los lactantes de piel negra, e infrecuente en las pieles más claras. La pigmentación se observa durante los primeros meses de vida, y va disminuyendo en intensidad durante el primer año.
Ayudas a la prescripción: interacciones de medicamentos
Introducción: La complejidad del proceso de utilización de los medicamentos es tal, que los errores asociados a su empleo pueden aparecer en uno, varios o incluso todos los pasos desde su prescripción a su dispensación. Con la introducción de ayudas sencillas en los programas de prescripción se puede mejorar de forma significativa la eficacia y la seguridad global de este proceso, y las ayudas sobre interacciones de los fármacos son las que más pueden influir en la calidad de la prescripción médica.
Objetivo: Desarrollar un programa de ayuda a la prescripción médica sobre interacciones de fármacos.
Método: Se seleccionan los medicamentos más utilizados en el área de cardiología pediátrica, revisándose para cada principio activo las interacciones fármaco-fármaco, alimento y producto medicinal.
Resultados: Se elabora una tabla con un total de 30 principios activos que recoge toda la información.
Discusión: Se estima que las interacciones pueden representar aproximadamente un 7% de todas las reacciones adversas en pacientes hospitalizados y el 1% del total de hospitalizaciones. Son muchas las posibles interacciones que pueden plantearse en la práctica clínica diaria, por lo que sólo hemos elegido las interacciones que se consideran más importantes por su potencial trascendencia clínica. La integración de esta información en el programa de prescripción electrónica constituye una ayuda a la prescripción médica, y puede contribuir a la disminución de la aparición de errores de medicación y problemas relacionados con los medicamentos.
Algunas tendencias en la ocupación del ocio en los jóvenes: ¿hacia un nuevo autismo?
La ocupación del tiempo libre de nuestros niños y jóvenes ha experimentado grandes cambios en las últimas dos décadas. El consumo televisivo se sitúa en >3 h/día. Más recientemente, este tiempo se ha visto desplazado hacia los videojuegos y consolas e Internet, con todas sus posibilidades. Por otra parte, la audición constante de música en reproductores individuales (mp3, mp4, i-pod) se presenta como una variable común en muchos jóvenes.
De estos cambios en los hábitos de vida (disminución consiguiente del tiempo de juego o de las actividades deportivas) se derivan consecuencias sobre el metabolismo (aumento del sedentarismo, obesidad, disminución de la forma física, etc.) o sobre la función fisiológica de algunos órganos (pérdida de audición para los sonidos agudos). Pero, además, tienen repercusiones sobre la capacidad de desarrollar habilidades sociales o sobre la comunicación. Estas dificultades en la interacción social o en la capacidad de comunicarse con los demás, o la aparición de patrones restringidos de comportamiento, actividades e intereses, es similar a muchas de las manifestaciones de los trastornos del espectro autista.
El pediatra tiene una tarea importante en el asesoramiento del uso de las nuevas tecnologías y también en el campo de la prevención de la aparición de «conductas aisladoras».
Revisión sistemática: tratamiento nutricional del cólico del lactante (y II)
Introducción: El cólico del lactante es un problema prevalente en el que se han ensayado a lo largo de la historia diversos tratamientos, ninguno de ellos definitivo. En los últimos años se han realizado nuevas propuestas terapéuticas, y han aparecido en el mercado las llamadas fórmulas anticólico (FAC), o confort. En el presente trabajo se realiza una revisión estructurada sobre el tratamiento nutricional del cólico del lactante, con el objetivo de integrar la información actual sobre este aspecto y establecer las pruebas existentes sobre la utilidad de distintas modalidades de tratamientvo nutricional.
Metodología: Se efectuó una revisión sistemática mediante búsqueda bibliográfica electrónica de los ensayos clínicos con diseño aleatorizado y controlado acerca del tratamiento nutricional del cólico del lactante.
Se realizó un análisis de las modificaciones en la composición de las FAC en relación con la fórmula adaptada convencional, según los datos proporcionados por los fabricantes del producto.
Resultados: Los 23 ensayos clínicos incluidos estudiaron seis modalidades de tratamiento nutricional: disminución de lactosa en la fórmula artificial, dieta hipoalergénica, adición de fibra, administración de soluciones azucaradas, probióticos y preparados fitoterápicos. Una proporción significativa de los trabajos analizados presenta problemas metodológicos, como un escaso número de pacientes, una alta tasa de pérdidas y sesgos de selección, que dificultan la extrapolación de sus resultados a la práctica clínica.
De las distintas intervenciones nutricionales, la exclusión de proteínas de leche de vaca en lactantes con fórmula artificial, la administración de preparados fitoterápicos y la dieta hipoalergénica extensa en la madre del lactante han demostrado tener algún grado de eficacia. Se necesitan estudios adicionales para verificar la eficacia de otras modalidades de tratamiento nutricional.
En Julio de 1958 «Acta Pediátrica Española» publicaba...
FIGURAS DE LA PEDIATRÍA
El doctor Crespo-Santillana
Saludo a la nueva Junta Directiva de la Sociedad de Pediatría de Madrid
ARTÍCULOS ORIGINALES
Trabajos doctrinales y casos clínicos
La sobremadurez en el recién nacido, por los doctores Luciano de la Villa y Antonio Fuertes
La terapéutica por clorpromazina en la prematuridad, por los doctores Luciano de la Villa y Antonio Fuertes
Síndromes neurológicos y psíquicos consecutivos a ciertas intervenciones quirúrgicas en el niño, por el doctor M. Schachter
Talón negro (hematoma subcórneo)
El talón negro es un proceso relativamente frecuente, caracterizado por la aparición de una lesión negra en la zona de apoyo del talón, que aparece en jóvenes deportistas. Está provocado por pequeños traumatismos que pueden dar lugar a hemorragias superficiales. Su interés patológico radica en la posible confusión con lesiones pigmentarias de estirpe melanocítica, lo que puede originar cierto grado de alarma. El diagnóstico es fácil de realizar y puede llevarse a cabo por el simple raspado de la lesión o mediante el examen con dermatoscopio.
Malaria en un niño procedente de adopción internacional
La malaria importada en la edad pediátrica es una enfermedad emergente en nuestro medio gracias al aumento de los viajes internacionales a países endémicos y a la llegada de niños inmigrantes o adoptados desde estas zonas. Describimos el caso de una niña adoptada procedente de Etiopía con parasitación por P. falciparum, que se encontraba asintomática a su llegada y sólo presentaba esplenomegalia como único hallazgo clínico. El diagnóstico y tratamiento precoz de esta enfermedad resulta fundamental para disminuir la morbimortalidad asociada, por lo que siempre debe descartarse en aquellos pacientes procedentes de área endémica.
Tuberculosis pulmonar complicada: a propósito de un caso docente
La tuberculosis es una enfermedad infecciosa que sigue siendo causa de una gran morbimortalidad infantil, tanto en los países en vías de desarrollo como en los desarrollados. En España, puede considerarse una enfermedad reemergente ligada a las nuevas formas de vida: viajes a zonas endémicas, alto grado de inmigración procedente de áreas de alta prevalencia, supervivencia a largo plazo de pacientes con alto riesgo de tuberculosis (inmunodeficientes congénitos y adquiridos), etc.
La población infantil actúa en muchas ocasiones como «centinela» de la infección o enfermedad tuberculosa dentro de una familia, grupo escolar, etc. De ahí la importancia de que los pediatras conozcan las formas típicas y atípicas de presentación de esta enfermedad, tengan un alto índice de sospecha sobre ella, y de forma inmediata procedan a realizar el diagnóstico y el tratamiento correctos, así como el estudio de contactos, que conllevaría cortar la cadena epidemiológica. El objeto de esta comunicación es revisar el tema a propósito de un caso de tuberculosis cavitada grave en un adolescente.
Hepatitis virales
En conjunto, las hepatitis virales suponen un importante problema mundial, con unas elevadas tasas de incidencia y prevalencia, que varían ampliamente según las distintas regiones.
Hasta ahora se han caracterizado cinco tipos distintos de virus causantes de hepatitis, con unas características clínicas y epidemiológicas diferentes. El A y el E se transmiten por vía fecal-oral y no producen enfermedad crónica. Por otro lado, el B, el D y el C se transmiten por vía parenteral y sexual principalmente, y pueden ocasionar, sobre todo en población pediátrica, una afectación crónica.
En el estudio inicial del niño inmigrante se debe incluir el cribado serológico de la infección por el virus de la hepatitis B. Aunque la vacunación frente a este virus es universal, no se suele realizar de modo adecuado la inmunoprofilaxis en el recién nacido.
La hepatitis A es endémica en países en vías de desarrollo. Suele afectar a niños que regresan a su país para visitar a amigos y familiares, por lo que es especialmente importante indicar la vacunación o la administración de gammaglobulina hiperinmune, según el caso, antes del viaje.
La incidencia de hepatitis C en general es baja en la infancia (<0,2%); sólo se debe realizar un cribado serológico en niños inmigrantes con factores de riesgo (antecedente de transfusión, hijos de madre con virus de la hepatitis C positivo, etc.).
Lúes congénita
La lúes, o sífilis, es la infección producida por Treponema pallidum y transmitida por contacto sexual, o de la madre al hijo por medio de transmisión vertical; esta última, la sífilis congénita, constituye el objeto de esta revisión. Presentamos un caso de sífilis congénita y posteriormente, en una breve discusión, incidimos en los factores epidemiológicos, diagnósticos y de tratamiento.
Infección por el virus de la inmunodeficiencia humana
Los casos de infección por el virus de la inmunodeficiencia humana (VIH) pediátricos en nuestro país han disminuido desde que se realiza la prevención de la transmisión vertical. Actualmente, los nuevos diagnósticos corresponden a fallos en dicha profilaxis, la mayoría de ellos porque ésta no se realizó correctamente, y también a niños que proceden de países con altas tasas de infección.
Se presenta un caso diagnosticado recientemente de infección por el VIH y se discute cómo debe realizarse la detección en el embarazo, cuándo se debe sospechar por la clínica dicha infección en la infancia y cuándo está indicado y cómo debe realizarse el tratamiento antirretroviral.
Vacunación en niños inmigrantes
En los últimos años se ha producido en España un aumento de la inmigración y de la adopción internacional. Los niños provienen generalmente de países en vías de desarrollo con coberturas vacunales bajas. En todo niño inmigrante o adoptado se debe realizar lo antes posible una evaluación de su estado vacunal y, en función de éste, completar las inmunizaciones, hasta adaptarlo al calendario de cada una de las comunidades autónomas (idealmente el recomendado por la Asociación Española de Pediatría).
Sólo hay que tener en cuenta las vacunas de las que se disponga de información fiable por escrito (teniendo en cuenta el número de dosis, el intervalo y la edad a la que se administraron), y considerar que toda «vacuna administrada es vacuna válida». En general, ante la duda, es preferible revacunar; alternativamente se pueden realizar determinaciones serológicas (difteria, tétanos, poliovirus 1-2 y 3, sarampión, rubéola y parotiditis). Se debe optar por pautas de vacunación rápida, inyectando el mayor número de dosis posibles a la vez y aprovechando cualquier visita para su administración. No se administran de modo sistemático en países en vías de desarrollo la vacuna heptavalente frente al neumococo, la vacuna Hib-conjugada ni la vacuna frente al meningococo C y frente a la varicela.
Aspectos nutricionales y del aparato digestivo de los niños inmigrantes
La mayoría de los niños que llegan a España generalmente no presentan las tasas de desnutrición del país de origen, aunque sí determinadas carencias nutricionales específicas (hierro, vitamina A) y raquitismo. En la actualidad, el hecho de pertenecer a una minoría étnica no debería ser un factor de riesgo para padecer desnutrición carencial y, con la excepción de los hijos de los inmigrantes recién llegados, el estado nutricional y el crecimiento de estos niños han de ser similares a los de los niños con el mismo nivel socioeconómico del país de destino.
Para un mejor control de los niños inmigrantes, los profesionales sanitarios deben conocer el estado nutricional de base (con la obtención de las distintas medidas antropométricas) y los aspectos genéticos y socioculturales, a fin de poder prevenir sus posibles alteraciones a largo plazo, ya que se están registrando importantes problemas de sobrepeso, especialmente en la segunda generación de esta población.
También repasamos la patología abdominal en el niño inmigrante desde el punto de vista sindrómico, para poder orientar el diagnóstico y el tratamiento. Aunque en general es similar a la observada en la población autóctona, debido a las características propias ambientales y a la carga genética de estos niños podemos hallar diferencias en la prevalencia de algunas enfermedades, aparte de las afecciones propias del trópico. Además, son frecuentes las patologías reactivas o de adaptación, que se expresan fundamentalmente con somatizaciones y síntomas vagos que indican una problemática relacional, y el dolor abdominal es el síntoma más común en estos niños.
Anemia falciforme
La anemia falciforme o drepanocitosis es una hemoglobinopatía estructural congénita que ha aumentado su prevalencia en España en la última década. Se caracteriza por una morfología del hematíe como una hoz, producida por la formación de polímeros de hemoglobina S en la célula. Esto ocasiona anemia hemolítica y trombosis. La clínica más frecuente es la siguiente: crisis de dolor óseo por vasooclusión, accidentes cerebrovasculares, infiltrados pulmonares, secuestro esplénico, asplenia funcional y disfunción de varios órganos. El tratamiento se basa en la educación sanitaria para el reconocimiento de los síntomas de alarma, la vacunación frente a gérmenes encapsulados, la profilaxis con penicilina, las transfusiones en situaciones seleccionadas, los estimuladores de la producción de hemoglobina fetal y, en casos graves, el trasplante de progenitores hematopoyéticos de hermano compatible.
Linfoma de Burkitt: el tumor pediátrico más frecuente en África
El linfoma de Burkitt supone casi un 40% de los linfomas no hodgkinianos en la infancia. Se reconocen tres variantes clínicas: asociado a inmunodeficiencia, esporádico y endémico, propio del África ecuatorial. Éste es el tumor pediátrico más frecuente en África ecuatorial, en cuya etiopatogenia actúan como cofactores la infección por el virus de Epstein-Barr y la malaria. Presenta diferencias clínicas y moleculares con los casos esporádicos, como la característica afectación de los huesos de la cara (principalmente de la mandíbula, seguida de la órbita), la mayor frecuencia de afectación del sistema nervioso central, de las glándulas salivales y del tiroides, y la menor frecuencia de infiltración de la médula ósea. El tratamiento se basa en quimioterapia combinada, y en el tratamiento inicial es crucial la prevención del síndrome de lisis tumoral. Los resultados del linfoma de Burkitt esporádico y endémico son superponibles, respecto a las tasas de respuesta, recaídas y supervivencia.
El niño africano: primera aproximación diagnóstica
La consulta de un niño procedente de una zona tropical es una situación cada día más habitual en nuestro país. Ante el aumento casi exponencial de la población inmigrante, es necesario tener en cuenta las patologías no endémicas en nuestro medio.
En este artículo se pretende enumerar las enfermedades infecciosas y tropicales propias de los niños africanos y ofrecer una primera aproximación diagnóstica de éstas en función de su sintomatología.
Manual práctico de nutrición en pediatría
Manual práctico de nutrición en pediatría
Coordinadores: M. Teresa Muñoz Calvo y Lucrecia Suárez Cortina
Sociedad de Pediatría de Madrid y Castilla-La Mancha y Comité de Nutrición de la Asociación Española de Pediatría.
Madrid: Ergón, 2007; 523 págs.
Manual práctico de nutrición en pediatría, o manual de nutrición práctica en pediatría, o manual de nutrición en pediatría práctica... Sea cual fuere tu nombre, ¡te estábamos esperando!
Hacía falta un manual así después de las interesantes experiencias que constituyeron los Tratados de nutrición en la infancia y en la adolescencia de los profesores A. Ballabriga y A. Carrascosa de 2006, ¡ya en su tercera edición!, o el Tratado de nutrición pediátrica, del profesor R. Tojo (2001) o incluso el amplio abordaje de los aspectos pediátricos en el excelente Tratado de nutrición del profesor Ángel Gil (2005).
Un libro de bolsillo, destinado tanto al pediatra de atención especializada como al de atención primaria. Y el desarrollo no podía ser más acertado: la mayoría de capítulos han sido elaborados de forma colaborativa por pediatras de ambos ámbitos de asistencia.
Las 523 páginas del libro están estructuradas en 30 capítulos y 7 anexos. Los primeros tres capítulos están dedicados a la valoración de los requerimientos nutricionales y del estado nutricional. Se sigue de un apartado amplio que abarca la nutrición en las distintas etapas de la infancia o en distintas situaciones no patológicas, como por ejemplo la alimentación del niño deportista o la alimentación en guarderías y colegios. Como una propuesta interesante, en un manual destinado a pediatras, se incluye un capítulo de aportes nutricionales en la mujer embarazada, entendiendo que la nutrición en el periodo fetal tiene repercusiones importantes en la vida del individuo.
Cinco capítulos se dedican a técnicas especiales de alimentación o a propiedades funcionales de los alimentos, incluyendo tanto aspectos de las técnicas de soporte nutricional como aspectos sobre los procesos tecnológicos de los alimentos o aditivos alimentarios. La parte más extensa de la obra se dedica al tratamiento nutricional en situaciones especiales: desnutrición, el niño que no come, enfermedades crónicas, déficit de vitaminas, patología digestiva, obesidad, diabetes mellitus, situaciones con riesgo cardiovascular o errores innatos del metabolismo. Un último capítulo sobre la educación nutricional pone al pediatra en el centro de las actividades de salud pública encaminadas a la mejora de la alimentación de nuestros niños y adolescentes. Se completa este volumen con 7 anexos con información relevante: tablas de percentiles, soluciones de rehidratación oral, fórmulas especiales, interacción de fármacos y alimentos o con la lactancia materna y una serie de consejos para los padres. Cada capítulo incluye una lista corta de bibliografía relevante, algunas de ellas comentadas.
Aunque la nutrición en la infancia y en la adolescencia constituyen una de las tareas primordiales de los pediatras, ha sido –y continúa siendo, en parte– una desconocida en la formación de posgrado. Manuales como el que ahora se publica pueden contribuir, por una parte, a concienciar a los pediatras más jóvenes del valor de la nutrición en la prevención y el tratamiento de las enfermedades y, por otra, a dar respuestas prácticas a los numerosos interrogantes que en la consulta diaria se nos plantean.
En Septiembre de 1958 «Acta Pediátrica Española» publicaba...
FIGURAS DE LA PEDIATRÍA
El doctor Selfa, de Valencia
ARTÍCULOS ORIGINALES
Trabajos doctrinales y casos clínicos
A propósito de un caso de estenosis hipertrófica de píloro, por los doctores Pérez Miró y Monsalve Pérez
Malformaciones congénitas de las vías naturales, por el doctor L. Gubern Salisachs
La pediatría social de los países comunistas, por el doctor Vicente Giménez
Estudio acerca de la contaminación del aire en un centro de prematuros, por el doctor Jorge Durich
Impétigo bulloso causado por «Staphylococcus aureus» resistente a meticilina
El impétigo es una infección cutánea superficial que ocurre sobre todo en la edad pediátrica, más frecuentemente por debajo de los 5 años de edad. Se clasifica en primario, que es el que tiene lugar sobre piel previamente sana, y secundario, que aparece en piel lesionada, principalmente tras un eccema. Existen dos tipos de impétigo: no bulloso, más frecuente, y bulloso. El agente causal predominante en todos los tipos de impétigo es Staphylococcus aureus. En los últimos años se ha descrito la emergencia de cepas de S. aureus resistentes a meticilina (SARM) como causantes de infecciones adquiridas en la comunidad, tanto leves como graves.
Se presenta el caso de un varón de 8 años que presenta lesiones ampollosas dolorosas de una semana de evolución en la región lumbar. Se recoge cultivo de las lesiones y se identifica el crecimiento de colonias de S. aureus con resistencia a meticilina.
Cefalohematoma calcificado persistente: a propósito de un caso
Los cefalohematomas son lesiones relacionadas con traumatismos obstétricos que afectan al 1,5-2,5% de los recién nacidos. Suelen desaparecer espontáneamente en pocas semanas. En el presente artículo se expone un caso de cefalohematoma calcificado persistente en un lactante de 5 meses, que no precisó tratamiento y que se resolvió cuando el paciente cumplió un año.
El absceso retrofaríngeo en la edad infantil
Introducción: Las infecciones del espacio retrofaríngeo tienen una baja incidencia. Suceden entre los 2 y los 5 años de edad normalmente. Son infecciones polimicrobianas, aunque dominan los estreptococos, estafilococos y gérmenes anaerobios. Producen dolor faríngeo, fiebre y disfagia, asociada a rigidez cervical. En la exploración se observa un abombamiento de la pared posterior faríngea.
Caso clínico: Presentamos el caso de un niño de 2 años que presentó un absceso de 11 cm3 de volumen, y otro de una niña de 4 años con un absceso de 8 cm3, por lo que se procede a la incisión y el drenaje del absceso por vía transoral de entrada.
Discusión: La tomografía computarizada (TC) cervical es la prueba diagnóstica de elección; permite realizar el diagnóstico en estadio temprano, así como la diferenciación entre celulitis y absceso.
El absceso retrofaríngeo rara vez se resuelve espontáneamente, por lo que es necesario administrar antibioterapia intravenosa y, si es mayor de 3 mL, un drenaje quirúrgico por vía transoral o externa mediante cervicotomía lateral.
Conclusión: La prueba estándar es la TC. Las complicaciones pueden ser muy graves.
Nutrición infantil y salud mental en el niño y en el adulto
En la última década, muchos estudios comprueban o sugieren que algunos de los llamados alimentos funcionales pueden proyectar su eficacia madurativa y preventiva del niño al adulto. Entre ellos, los ácidos grasos poliinsaturados de cadena larga (LC-PUFA) desempeñan un importante papel tanto en el desarrollo del sistema nervioso como en la prevención de diferentes enfermedades neuropsiquiátricas. Otros nutrientes, como los nucleótidos, los oligosacáridos, los gangliósidos, el colesterol o los micronutrientes (hierro, cinc, ácido fólico), también están involucrados directa o indirectamente en la salud mental del propio niño o el adulto, y se describen más someramente en este artículo de revisión.
Pioderma facial
El pioderma facial o rosácea fulminans es una enfermedad dermatológica poco frecuente que afecta a la cara y cursa con lesiones muy inflamatorias que pueden dejar cicatrices residuales. El tratamiento debe instaurarse de forma precoz; los corticoides orales y la isotretinoína oral son los fármacos más empleados. Presentamos el caso de una paciente adolescente que evolucionó favorablemente con este tratamiento.
Uso de dexametasona en la meningitis bacteriana pediátrica y neonatal
La meningitis bacteriana es una infección grave por la alta incidencia de fallecimiento y por las secuelas permanentes que induce. Existen numerosas dudas y contradicciones en cuanto a las situaciones en las que deberían utilizarse corticosteroides como terapia asociada a los antibióticos, así como con la dosis y duración idónea de su administración. El objetivo de esta revisión sistemática es actualizar los conocimientos sobre las indicaciones del uso de dexametasona en meningitis piógenas, así como la pauta de tratamiento más adecuada.
Aumento de la hospitalización por varicela y de sus graves complicaciones en los niños de la isla de Mallorca
El objetivo de este estudio es determinar las tasas de infección, hospitalización y complicaciones de la varicela en la isla de Mallorca. Para estudiar la incidencia de esta infección, revisamos retrospectivamente los casos notificados a la Red de Vigilancia Epidemiológica durante el periodo 1982-2005. Las tasas de hospitalización se obtuvieron de los registros de alta de los pacientes ingresados en los tres hospitales públicos de la isla de Mallorca, durante los periodos 1995-2000 y 2001-2005. Las tasas de admisión y las complicaciones se estudiaron también específicamente en el hospital de referencia.
La incidencia de varicela descendió desde 994/100.000 habitantes en el periodo 1982-1989, a 521 en el periodo 2001-2005 (p <0,0001). Globalmente, la tasa de hospitalización por varicela aumentó significativamente desde 10,6/100.000 habitantes menores de 15 años (o 1,62/1.000 casos de varicela) en el periodo 1995-2000 a 26,4/100.000 (o 3,54/1.000 casos) en el periodo 2001-2005 (p <0,0001). La edad media de los pacientes fue de 2,8 años, y la mortalidad fue nula. Las complicaciones más frecuentes en los 147 niños ingresados en el hospital de referencia fueron: celulitis (13%), impétigo (10,2%), neumonía (9,5%), convulsiones (8,8%) y artritis (4,8%). Se observó un aumento de la prevalencia de sepsis en pacientes hospitalizados por varicela (2/46 frente a 9/101; p= 0,3).
Aunque parece haber un descenso de los casos de varicela en la isla de Mallorca, observamos un incremento de la hospitalización y de complicaciones graves por esta infección. La vacuna contra la varicela a la edad de 12-18 meses probablemente reduciría esta tendencia.
Trombosis arteriales y venosas en la infancia (I): incidencia, etiopatogenia y diagnóstico
La incidencia de las trombosis en la población infantil es de 0,07/10.000; en el 5,3/10.000 de los casos, durante el ingreso hospitalario, y en el 2,4% de los casos durante la admisión en la Unidad de Cuidados Intensivos. En el primer año de vida, la prevalencia de tromboembolia venosa es 40 veces superior a la de otras edades de la infancia. La trombosis espontánea, sin causa etiológica que la justifique, precisa uno o más factores de riesgo protrombóticos para su aparición. Existen factores de riesgo congénitos, debidos a alteraciones de la hemostasia, que suponen un riesgo mayor si se asocian a otros factores genéticos. El correcto diagnóstico mediante el estudio de los signos clínicos y de las técnicas de diagnóstico no invasivas e invasivas, así como de la pruebas de laboratorio, en las que el dímero-D ha demostrado una alta sensibilidad, son imprescindibles antes de iniciar cualquier tipo de tratamiento antitrómbico.
En Diciembre de 1957 «Acta Pediátrica Española» publicaba...
FIGURAS DE LA PEDIATRÍA
El doctor Prandi Farrás, de Barcelona
ARTÍCULOS ORIGINALES
Trabajos doctrinales y casos clínicos
Las hemoglobinas normales y patológicas del lactante y el niño, por los doctores Giraud, Orsini y Le Poullain
Estudio de las lipoproteínas y glucoproteínas en el recién nacido, por electroforesis en papel, por los profesores Galdóy M. Cruz-Hernández, y el doctor B. Esteban
Multiplicidad de las malformaciones congénitas. Exposición casuística, por el doctor L. Fiuza Pérez
Problemas de nutrición en la población escolar madrileña, por el doctor A. Serigó
Crónica del Curso de Pediatría Social. París, 1957, por el doctor E. Burgos
Mortalidad infantil por anomalías congénitas: beneficio de la intervención quirúrgica, por el doctor J. Garrido Lestache
Nevo comedoniano. Respuesta satisfactoria al ácido retinoico
Sr. Director:
El nevo comedoniano es una malformación folicular constituida por múltiples lesiones con aspecto de comedones de distintos tamaños, agrupados linealmente o en parches, que se distribuyen habitualmente de forma unilateral, aunque se han descrito algunos casos de distribución bilateral1.
Un niño de 6 años, sin antecedentes de interés, acude a nuestra consulta por presentar desde hace un año y medio unas lesiones en la zona posterolateral derecha del cuello, ocasionalmente pruriginosas.
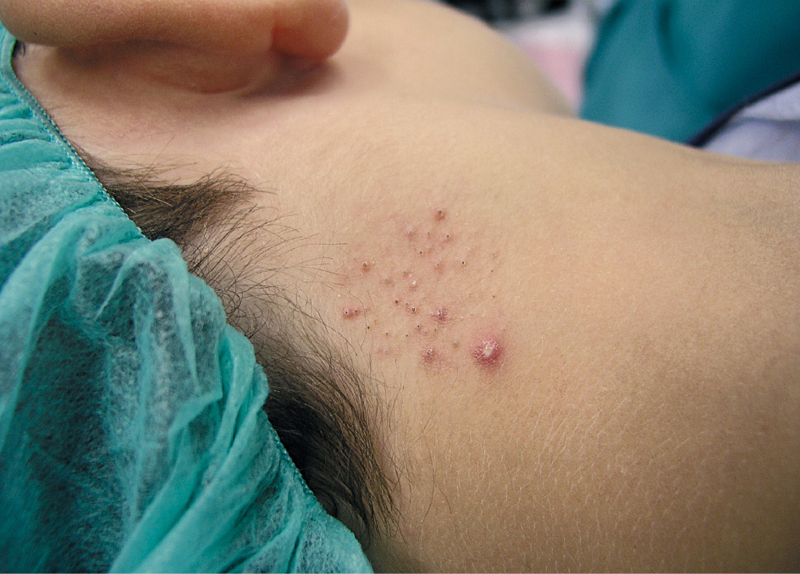
En la exploración dermatológica se observan, sobre un área bien delimitada de aproximadamente unos 3 cm de diámetro, múltiples pápulas centradas por tapones hiperqueratósicos similares a comedones abiertos, con alguna pústula aislada (figura 1).
No presenta antecedentes familiares de lesiones similares.
Ante la sospecha de nevo comedoniano, se realizó una biopsia que confirmó dicho diagnóstico.
Se inició tratamiento con ácido retinoico al 0,025% una vez al día, desapareciendo gran parte de las lesiones en aproximadamente 2 meses.
El nevo comedoniano puede aparecer en la pubertad o estar presente desde el nacimiento. Suelen ser lesiones asintomáticas y generalmente unilaterales, con preferencia por ciertas zonas, como la cara, el cuello, el tronco o las extremidades superiores1,2. Su tamaño es variable y oscila desde unos pocos centímetros a lesiones extensas que afectan a un hemicuerpo entero2.
Su etiopatogenia no está clara, pero parece tratarse de un mosaicismo genético2.
Es necesario realizar un diagnóstico diferencial con la acné vulgar y neonatal. La distribución unilateral y la persistencia del nevo comedoniano lo diferencian de estas entidades. Otras afecciones con las que se puede confundir son la cloracné, los comedones disqueratósicos familiares, el nevo ostial poroqueratósico ecrino, la enfermedad de Darier lineal y el nevo sebáceo2.
Dado que es una lesión benigna no necesita tratamiento, salvo por motivos estéticos o para evitar infecciones secundarias.
La extirpación quirúrgica es una opción terapéutica en las lesiones localizadas y de escasa extensión2.
Otros tratamientos que se pueden valorar, aunque con respuesta variable, son los siguientes: láser de CO2, dermoabrasión, derivados de la vitamina D3 tópicos (calcipotriol, tacalcitol)3, agentes queratolíticos, como el ácido salicílico, los hidroxiácidos alfa y las cremas de lactato amónico al 12%4. Otra opción, que en el paciente del caso expuesto tuvo una respuesta muy satisfactoria, es el ácido retinoico5 tópico, en sus distintas concentraciones.
Bibliografía
- Izquierdo MJ, Requena C, Requena L. Nevo comedoniano. En: Neoplasias anexiales cutáneas. Madrid: Grupo Aula Médica, 2004; 211-214.
- Monteagudo Sánchez B, Ginarte Val M, León Muiños E, Vázquez Golpe R, Varela Iglesas A. Nevo comedoniano. An Pediatr (Barc). 2006; 65(2): 171-172.
- Wakahara M, Kiyohara T, Kumakiri M, Kuwahara H, Fujita T. Bilateral nevus comedonicus: efficacy of topical tacalcitol ointment. Acta Derm Venereol. 2003; 83: 51.
- Bordel MT, Miranda A. Nevo comedoniano unilateral: eficacia tras el tratamiento con lactato amónico al 12%. Actas Dermosifiliogr. 2006; 97: 150.
- Decherd JW, Mills O, Leyden JJ. Naevus comedonicus treatment with retinoic acid. Br J Dermatol. 1972; 86: 528-529.
Síndrome compartimental tras una mordedura de víbora
Presentamos el caso de un niño de 12 años de edad que, estando en un campamento de verano, sufre una mordedura de víbora que le desencadena un síndrome compartimental grave.
Diagnóstico perinatal de rabdomiomas intracardiacos y esclerosis tuberosa
Los tumores cardiacos son poco comunes, y los más frecuentes son los rabdomiomas. Su diagnóstico puede realizarse durante la época fetal. La evolución es variable: algunos son asintomáticos y otros se manifiestan por una obstrucción al flujo o en forma de arritmia. Este tipo de tumores puede ser la clave diagnóstica de una esclerosis tuberosa. Dado el amplio espectro en su presentación, mostramos dos casos, uno de diagnóstico prenatal y otro a partir de un hallazgo en la ecocardiografía.
Anemia de células falciformes y varicela en gemelo de un mes de edad
Presentamos un caso de anemia de células falciformes (ACF) en un varón de raza negra de un mes de edad, gemelo heterocigoto de una niña, que presenta una crisis hemolítica y/o aplásica en el periodo de estado de una varicela. Acude a urgencias de nuestro hospital por una posible crisis de atragantamiento, donde se detecta una anemia con 7 g/dL de hemoglobina (Hb), que desciende en las siguientes 48 horas a 6,8 g/dL. Tras la corrección de la anemia con 60 mL de concentrado de hematíes, el nivel de Hb asciende a 13 g/dL. En la exploración no presenta esplenomegalia y la Hb F es del 85,2%. Por tanto, consideramos el papel que podría desempeñar la infección por el virus de la varicela en la producción de la crisis aplásica y la falta de protección de la Hb F, pese a sus altos valores.
Administración de hierro intravenoso en niños. Aspectos prácticos
El hierro es un nutriente esencial con un papel fisiológico muy importante para la vida. Su déficit ocasiona la anemia ferropénica, la enfermedad hematológica más frecuente en la infancia, cuyo tratamiento se fundamenta, por un lado, en la corrección de la causa que la origina y, por otro, en la administración de suplementos de hierro. En algunos casos en que el tratamiento con hierro oral no es posible, debe recurrirse a su administración parenteral. Esta vía de administración permite aportar el hierro más rápidamente, lo que supone mayor rapidez en la recuperación de la anemia, con los consiguientes beneficios. La disponibilidad de un preparado como el hierro sacarosa, con muy buen perfil de seguridad y eficacia, justifica su empleo en pacientes pediátricos ferropénicos.
Dermatitis de Berloque en la infancia
La dermatitis de Berloque es una fotodermatosis que requiere una anamnesis temporal cuidadosa: el antecedente de la aplicación de la sustancia en la superficie corporal y la exposición solar posterior nos dan la clave para su correcto diagnóstico.
Aunque habitual para el dermatólogo, no es un cuadro clínico común en el paciente pediátrico y debe incluirse en el diagnostico diferencial de las dermatitis de contacto, incluso en casos de maltrato infantil.
Estudio de 6 años sobre neumonía por «Mycoplasma pneumoniae» en un hospital terciario pediátrico
Objetivos: Determinar la importancia de la incidencia de Mycoplasma pneumoniae en la etiología de las neumonías adquiridas en la comunidad en pacientes pediátricos de edades comprendidas entre los 6 meses y los 16 años.
Metodología: Se recogieron, de forma prospectiva, todos los casos diagnosticados de neumonía por Mycoplasma pneumoniae, en el servicio de urgencias del departamento de pediatría del Hospital Materno Infantil Universitario «Gregorio Marañón» de Madrid, desde el 1 de mayo de 1995 hasta el 31 de agosto del 2001. En todos los pacientes se recogieron datos epidemiológicos (edad, sexo, incidencia familiar, asistencia a colegio o guardería, fecha del diagnóstico...), clínicos (síntomas y exploración física en el momento del diagnóstico y seguimiento en la consulta de Infecciosas), hematométricos, serológicos y radiológicos.
Resultados: Desde el 1 de mayo de 1995 hasta el 1 de septiembre de 2001 se diagnosticaron en el servicio de urgencias infantiles un total de 715 neumonías en pacientes con edades comprendidas entre los 6 meses y los 16 años. Se confirmó el diagnóstico de neumonía por M. pneumoniae en 240 casos (34%). Casi la mitad de los pacientes tenían una edad igual o inferior a los 5 años de edad. La mayoría de las neumonías fueron tratadas ambulatoriamente; sólo un 4,5% de los pacientes diagnosticados de neumonías por M. pneumoniae precisaron ingreso frente a un 23,15% en el grupo de neumonías por otros agentes etiológicos (p <0,05). La distribución mensual de las neumonías por M. pneumoniae presenta dos picos epidémicos (de diciembre de 1997 a agosto de 1998 y de febrero de 2001 a agosto de 2001). Todos los pacientes diagnosticados evolucionaron satisfactoriamente.
Conclusión: M. pneumoniae constituye una causa frecuente de neumonía ambulatoria en la comunidad, incluso en pacientes menores de 5 años, y presenta una excelente evolución tras tratamiento ambulatorio.
Chagas vertical: una realidad en España
La enfermedad de Chagas, o tripanosomiasis americana, es una patología endémica en amplias regiones de América, desde el sur de Estados Unidos hasta el sur de Argentina y Chile.
En la actualidad, el peligro de contraer la enfermedad no sólo está en Latinoamérica. Debido a los movimientos migratorios de la población de dichas regiones, la infección está llegando a ser un problema mundial.
Es necesario actualizar los conocimientos sobre esta enfermedad emergente, que ya es una realidad en nuestro medio, mediante una revisión bibliográfica.
Las mujeres embarazadas infectadas pueden transmitir la enfermedad por vía vertical o transplacentaria a su hijo; esta última es la vía que nos interesa, por dar lugar al Chagas congénito. En áreas endémicas la prevalencia de transmisión por vía transplacentaria oscila entre el 2 y el 12%.
Es importante solicitar las pruebas serológicas a las mujeres embarazadas provenientes de áreas endémicas y, en caso de ser positivas, realizar el diagnóstico precoz de sus hijos y el posterior tratamiento durante el primer año de vida.
Hipercolesterolemia en la infancia y la adolescencia
La enfermedad cardiovascular continúa siendo la primera causa de morbimortalidad en los países industrializados en la edad adulta, pero de los datos disponibles podemos afirmar que las alteraciones iniciales, poco evidentes pero con gran significado futuro, se inician en la mayoría de las personas durante la infancia.
El objeto de este trabajo es una puesta al día de esta epidemia, contemplando inicialmente el metabolismo complejo del colesterol, la evaluación del riesgo futuro y las medidas que deben adoptarse, tanto desde un punto de vista preventivo como de enfoque de esta situación, así como de los tratamientos farmacológicos de que disponemos, sus indicaciones y sus riesgos, que no deben ser minusvalorados.
Las medidas preventivas que se pueden aconsejar en la población pediátrica sin factores de riesgo no son fáciles de tomar y, hasta que no se disponga de un marcador biológico, el problema continuará sin resolverse. Distinto es el problema cuando se conocen los factores de riesgo por la historia familiar, en cuyo caso, además de medidas dietéticas y ejercicio regular, puede ser aconsejable el uso de estatinas, pero sin olvidar sus riesgos potenciales y la experiencia escasa que existe todavía al respecto.
Síndrome de activación de macrófagos: una entidad que debe considerarse dentro de la linfohistiocitosis hemofagocítica
Sr. Director:
He leído con interés el caso clínico publicado en su revista diagnosticado de síndrome de activación macrofágica (SAM) y artritis juvenil de comienzo sistémico (AJS)1, del que quisiera hacer varias consideraciones.
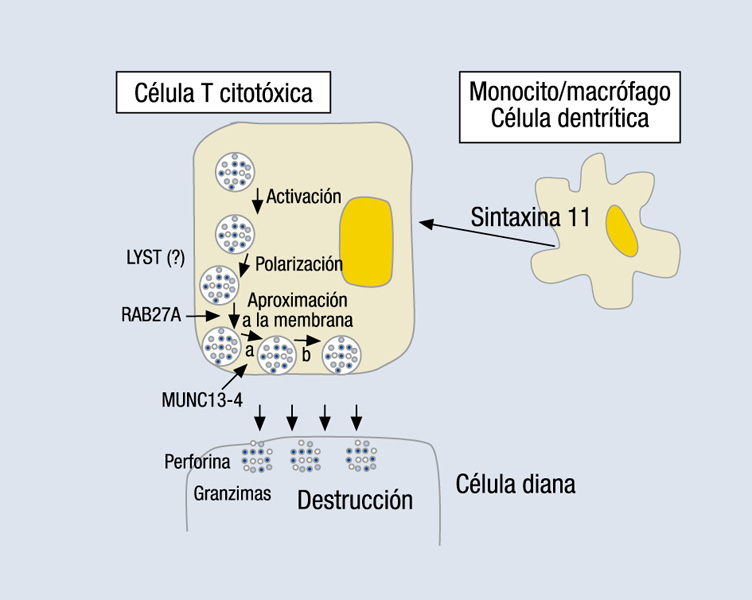
 Como comentan los propios autores, el SAM presenta características clínicas similares a la linfohistiocitosis hemofagocítica (LHH) y, de hecho, no debería considerarse fuera de dicha entidad2, como recomienda la propia Sociedad Histiocitaria3 y alguno de los autores citados en el artículo4. La LHH es un cuadro grave que puede ser de origen genético (familiar) o adquirido desencadenado por infecciones, procesos malignos, algunas metabolopatías y también por enfermedades autoinmunitarias o reumatológicas (el llamado SAM)3-6. Los criterios diagnósticos propuestos por la Sociedad Histiocitaria para la LHH3 se muestran en la tabla 1. La patogenia común es la presencia de un defecto adquirido o congénito de la función NK y citotóxica, que impide la destrucción de la célula infectada manteniéndose la actividad inflamatoria de las células histiocitarias y linfocitos T con persistencia de niveles altos de citocinas, que son las que originan el cuadro clínico5,6. Tanto las formas familiares como las adquiridas pueden estar desencadenadas por infecciones, y las más frecuentes son las producidas por virus del tipo herpes, como el virus de Ebstein-Barr o el de la varicela6, como en el caso publicado1. Por ello, es difícil diferenciar ambos cuadros si no hay antecedentes familiares, ya que son clínica y anatomopatológicamente indistinguibles3,5, y debe sospecharse la posibilidad de un origen genético cuando la enfermedad se manifiesta en el primer año de vida5. El descubrimiento de mutaciones en varios genes implicados ha demostrado que también hay casos familiares que se manifiestan tardíamente, hasta la cuarta década de la vida5,6. Los genes descritos afectan al gen de la perforina (PRF1), al MUNC13-4 (o UNC13D), a la sintaxina 11 (STX11) y, al menos, a otro gen desconocido localizado en el cromosoma 93,5. Todos ellos, junto con los genes afectados en la enfermedad de Griscelli, en la enfermedad de Chediag-Higashi o en el síndrome linfoproliferativo ligado al cromosoma X, afectan a la capacidad de liberación y el tráfico de gránulos citolíticos desde la célula efectora a la célula diana5, como se muestra en la figura 1.
Como comentan los propios autores, el SAM presenta características clínicas similares a la linfohistiocitosis hemofagocítica (LHH) y, de hecho, no debería considerarse fuera de dicha entidad2, como recomienda la propia Sociedad Histiocitaria3 y alguno de los autores citados en el artículo4. La LHH es un cuadro grave que puede ser de origen genético (familiar) o adquirido desencadenado por infecciones, procesos malignos, algunas metabolopatías y también por enfermedades autoinmunitarias o reumatológicas (el llamado SAM)3-6. Los criterios diagnósticos propuestos por la Sociedad Histiocitaria para la LHH3 se muestran en la tabla 1. La patogenia común es la presencia de un defecto adquirido o congénito de la función NK y citotóxica, que impide la destrucción de la célula infectada manteniéndose la actividad inflamatoria de las células histiocitarias y linfocitos T con persistencia de niveles altos de citocinas, que son las que originan el cuadro clínico5,6. Tanto las formas familiares como las adquiridas pueden estar desencadenadas por infecciones, y las más frecuentes son las producidas por virus del tipo herpes, como el virus de Ebstein-Barr o el de la varicela6, como en el caso publicado1. Por ello, es difícil diferenciar ambos cuadros si no hay antecedentes familiares, ya que son clínica y anatomopatológicamente indistinguibles3,5, y debe sospecharse la posibilidad de un origen genético cuando la enfermedad se manifiesta en el primer año de vida5. El descubrimiento de mutaciones en varios genes implicados ha demostrado que también hay casos familiares que se manifiestan tardíamente, hasta la cuarta década de la vida5,6. Los genes descritos afectan al gen de la perforina (PRF1), al MUNC13-4 (o UNC13D), a la sintaxina 11 (STX11) y, al menos, a otro gen desconocido localizado en el cromosoma 93,5. Todos ellos, junto con los genes afectados en la enfermedad de Griscelli, en la enfermedad de Chediag-Higashi o en el síndrome linfoproliferativo ligado al cromosoma X, afectan a la capacidad de liberación y el tráfico de gránulos citolíticos desde la célula efectora a la célula diana5, como se muestra en la figura 1.
Los casos de SAM aparecen, en general, más tarde, y suelen ser menos graves que los congénitos o que los adquiridos tras infecciones, aunque también pueden ser mortales5,7. En el caso publicado llama la atención que se hizo el diagnóstico de AJS a los 9 meses, por un síndrome febril prolongado con hepatosplenomegalia sin afectación articular, en el que había una elevación de la proteína C reactiva (PCR) y del factor reumatoide. No se descarta que en realidad se tratara de una primera manifestación de LHH, pues desconocemos si había otros parámetros analíticos que lo apoyaran, y el hecho de que en ese primer momento la punción de médula ósea fuera normal no lo descarta, ya que la LHH es un cuadro progresivo y la hemofagocitosis puede no demostrarse al principio5,6. Los corticoides actúan como linfocidas e inhiben la expresión de citocinas y quimiocinas5, que pueden haber abortado el cuadro inicial, siendo insuficiente para la curación del mismo, sobre todo si, por la precocidad de la aparición de la clínica, se trata de un caso familiar. Es interesante el hecho de que 4 días después del comienzo de la fiebre por la varicela presentara esplenomegalia en la exploración, un dato absolutamente atípico en una varicela no complicada. La sospecha diagnóstica de LHH debe ser precoz, dado que el cuadro puede ser fulminante si no se instaura un tratamiento adecuado, como ocurrió en este caso. La Sociedad Histiocitaria ha elaborado un protocolo de tratamiento3 en el que se emplea dexametasona, ciclosporina y etopósido (disponible en www.histio.org/society/protocols). Presenta pequeñas variaciones frente a uno inicial, que ha demostrado su eficacia tanto en las formas genéticas como en las adquiridas8, incluido el SAM. La remisión completa tras el tratamiento farmacológico sería suficiente para las formas adquiridas, pero la reaparición o la reactivación del cuadro, o el diagnóstico de una forma genética (por historia familiar o diagnóstico genético), necesita además el trasplante de células progenitoras hematopoyéticas para su curación definitiva3,8.
Janka5 reconoce que es difícil diferenciar el SAM de otras formas de LHH cuando falta la artritis, y sugiere que en su experiencia una PCR elevada (como tenía la paciente) apoyaría el primer diagnóstico. Aunque el tratamiento menos agresivo del SAM puede ser eficaz, sobre todo en niños mayores sin un aparente factor desencadenante, como comentan los autores1, en este caso grave, desarrollado tras la varicela, debería haberse empleado el tratamiento propuesto por la Sociedad Histiocitaria si la entidad se hubiera sospechado y diagnosticado antes de su fallecimiento.
Bibliografía
- Aleo Luján E, Gil López C, Balboa de Paz F, Kilmurray GL, Pérez Rodríguez O, Rubial Francisco JL. Síndrome de activación de macrófagos y artritis juvenil de inicio sistémico. Acta Pediatr Esp. 2007; 65: 304-308.
- Remanan AV, Baildam EM. Macrophage activation syndrome is hemophagocytic lymphohistiocytosis-need for the right terminology. J Rheumathol. 2002; 29: 1.105.
- Henter JI, Horne AC, Aricò M, Egeler RM, Filipovich AH, Imashuku S, et al. HLH-2004: diagnostic and therapeutic guidelines for hemophagocytic lymphohistiocytosis. Pediatr Blood Cancer. 2007; 48: 124-131.
- Ravelli A. Macrophage activation syndrome. Curr Opin Rheumatol. 2002; 14: 548-552.
- Janka GE. Familial and acquired hemophagocytic lymphohistiocytosis. Eur J Pediatr. 2007; 166: 95-109.
- Filipovich AH. Hemophagocytic lymphohistiocytosis and related disorders. Curr Opin Allergy Clin Immunol. 2006; 6: 410-415.
- Chen HH, Kuo HC, Wang L, Yu H, Shen JM, Kwang KP, et al. Childhood macrophage activation syndrome differs from infection-associated hemophagocytosis syndrome in etiology and outcome in Taiwan. J Microbiol Immunol Infect. 2007; 40: 265-271.
- Henter IH, Samuelsson-Horne AC, Aricò M, Egeler RM, Elinder G, Filipovich AH, et al. Treatment of hemophagocytic lymphohistiocytosis with HLH-94 immunochemotherapy and bone marrow transplantation. Blood. 2002; 100: 2.367-2.373.
En Noviembre de 1957 «Acta Pediátrica Española» publicaba...
FIGURAS DE LA PEDIATRÍA
El profesor Lelong, de París
ARTÍCULOS ORIGINALES
Trabajos doctrinales y casos clínicos
Disostosis membranosa con hipervitaminosis A y D, por el doctor A. Valls
Secuencias psíquicas del hijo rechazado, por el doctor J. Moragas
La selección madre-hijo, por el doctor J. Molina
La agresividad infantil como problema, por el doctor F. Mendiguchía
Aportación personal de un caso de adamantinoma en la infancia, por el doctor Andrés G. Meneses Pardo
La hipoproteinemia idiopática del niño, por el doctor J.A. de Paz Garnelo
Eritema exudativo multiforme, forma «minor»
Presentamos el caso de un paciente varón, de 6 años de edad, visitado en el Servicio de Urgencias de Pediatría del Hospital General Universitario «Gregorio Marañón» de Madrid (HGUGM), por presentar desde hacía 2-3 días un proceso catarral, con fiebre de 38-39 ºC, aftas bucales, con descamación en los labios, que se acompañaba en las últimas horas de un exantema maculo-papulo-pruriginoso generalizado, más intenso en los miembros superiores e inferiores, con artralgias y artritis interfalángicas.
En la exploración que se realiza en el servio de urgencias, se aprecia un eritema urticariforme muy significativo, más notorio en los miembros, con evolución de las lesiones cutáneas a lesiones en diana.
Seudoquiste pancreático en dos pacientes menores de 24 meses
El seudoquiste pancreático se produce como consecuencia de un proceso inflamatorio y/o traumático; su contenido es estéril y rico en enzimas pancreáticas. Habitualmente, debe sospecharse ante una elevación persistente de la amilasa, puesto que suele ser asintomático, salvo complicaciones. El 40-50% de los seudoquistes se resuelven espontáneamente, y deben drenarse los mayores de 6 cm y/o que persistan más de seis semanas, dado el riesgo de complicaciones.
Presentamos los casos de dos pacientes menores de 24 meses con seudoquistes pancreáticos secundarios a pancreatitis agudas: el primero con una evolución típica y el segundo con una elevación aislada de la lipasa.
Hipertransaminemia persistente sin clínica digestiva: ¿hay que pensar en celiaquía?
Se presenta el caso de un lactante de 2 años, con estancamiento de peso en los últimos 4 meses, advertido en la revisión efectuada por su pediatra, sin que mostrara clínica intestinal evidente y con resultado de analítica de hipertransaminemia elevada (GOT 173, GPT 425), sin otros datos clínicos ni analíticos de hepatopatía, y serología de virus hepatótropos negativa.
Ante la persistencia de la hipertransaminemia (en el control efectuado a los 20 días se obtuvieron valores similares), se amplía el estudio, con resultados de AAG 1/80 (positivo >1/20) y AAT 30 UA/mL (positivo >12); seguidamente, se realiza una biopsia intestinal, con resultado de atrofia vellositaria subtotal, linfocitosis e hiperplasia de criptas. Tras 2 meses de dieta estricta sin gluten, se repitió la analítica, y se constató una normalización total de las transaminasas.
Síndrome de aracnodactilia con dolicostenomelia y contracturas (síndrome de Beals)
El síndrome de Beals es una enfermedad hereditaria del tejido conectivo. Tiene un carácter autosómico dominante. Fenotípicamente, es parecido al síndrome de Marfan, pero menos grave. Se caracteriza por aracnodactilia, dolicostenomelia, cifoscoliosis, contracturas múltiples congénitas, alteraciones de los pabellones auriculares, hipoplasia de los músculos de la pantorrilla y cardiopatía. El gen responsable de este síndrome (5q-23-31) codifica una proteína, la fibrilina 2, componente de las microfibrillas de elastina. La evolución de las contracturas es hacia la mejoría, pero la cifoscoliosis puede ser progresiva. Describimos un nuevo caso y revisamos la bibliografía médica.
Síndrome de Bannayan-Riley-Ruvalcaba: a propósito de un caso
Introducción: El síndrome de Bannayan-Riley-Ruvalcaba (SBRR) es una enfermedad autosómica dominante, caracterizada por lesiones hamartomatosas que afectan a diversos tejidos. Los tumores más habituales son los lipomas y las malformaciones vasculares (hemangiomas). El SBRR produce macrosomía y macrocefalia en la mayoría de los pacientes, retraso mental de leve a grave y, ocasionalmente, tiroiditis autoinmunitaria. Con frecuencia, el SBRR se asocia a mutaciones en la línea germinal del gen de supresión tumoral PTEN.
Caso clínico: Niña de 12 años que acude a nuestro servicio de neuropediatría por presentar hemangioma lingual, macrosomía sin macrocefalia, tiroiditis autoinmunitaria y problemas escolares, que llevaron al diagnóstico de trastorno por déficit de atención con hiperactividad. Ante la sospecha de un SBRR, se realiza un test genético, en el que detecta una mutación intrónica del gen PTEN.
Conclusiones: PTEN es un supresor tumoral. Mutaciones en diferentes splice sites de PTEN producen deleciones de diferentes exones, que conducen a resultados distintos. Los pacientes que presentan una mutación tipo intrón profundo (deep intronic mutation) parecen tener un fenotipo más leve que los que muestran otros tipos de mutaciones. La presencia de macrocefalia no es imprescindible para el diagnóstico. Dado el riesgo que presentan estos pacientes de desarrollar tumores, es importante realizar un diagnóstico adecuado.
Probióticos para el binomio madre-hijo (y II)
En esta parte del trabajo se revisan los conocimientos actuales sobre aplicación de probióticos en niños prematuros y en mujeres embarazadas y lactantes. Los estudios de administración de cepas probióticas a prematuros son relativamente escasos, aunque, en general, indican que la incidencia de enterocolitis necrosante y septicemia es menor en prematuros a los que se les administra un probiótico. En cualquier caso, las cepas empleadas hasta la fecha han demostrado ser seguras en estos hospedadores tan sensibles a las enfermedades infecciosas. De hecho, el éxito del método de la madre canguro, en el que los prematuros están expuestos a las bacterias comensales y probióticas de la leche materna, confirma los beneficios del contacto precoz con este tipo de agentes. En cuanto a los niños con síndrome de intestino corto, resulta sorprendente que la administración de ciertos lactobacilos productores de D-lactato no sólo no es perjudicial, sino que conduce a una remisión de la sintomatología más rápida que cuando se administran cepas probióticas que exclusivamente producen L-lactato. Finalmente, las mujeres embarazadas y los lactantes constituyen una población en la que la administración de probióticos podría resultar de particular interés, dado que la modulación de sus microbiotas digestiva, mamaria y urogenital puede tener una importante repercusión sobre la salud del binomio madre-hijo.
Aplasia cutánea congénita
La aplasia cutánea congénita (ACC) es una rara alteración caracterizada por la ausencia congénita de epidermis, dermis y, en ocasiones, de los tejidos subyacentes. La forma más frecuente afecta al vértex, y se puede presentar de forma aislada o asociada a otras malformaciones. Fue descrita por primera vez por Cordon, en 1767, y desde entonces se han documentado más de 500 casos. La frecuencia se ha estimado en torno a 3/10.000 recién nacidos. No hay predilección por la raza ni el sexo. La etiopatogenia no está clara. La manifestación clínica habitual es una lesión oval o circular, solitaria, sin pelo, bien delimitada, de 0,5-2 cm, y localizada en el vértex. Al nacer, las lesiones pueden estar cicatrizadas o presentarse como erosiones o incluso úlceras profundas, con riesgo de afectar a las meninges o la duramadre. La mejor opción terapéutica dependerá del tamaño del defecto.
Síndrome inv dup(15): a propósito de dos casos
Introducción: La región cromosómica 15q11q13 es inestable debido a la presencia de elementos de ADN repetidos. En esta región pueden producirse muchas reordenaciones estructurales que habitualmente afectan a la región crítica Prader-Willi-Angelman (RCPWA), como deleciones, translocaciones, inversiones y cromosomas marcadores supernumerarios (CMS), formados por la duplicación invertida de la región proximal del cromosoma 15.
En este artículo, se describen clínicamente los casos de 2 pacientes afectados del síndrome producido por una duplicación invertida del cromosoma 15, que se denomina inv dup(15) o idic(15).
Casos clínicos: Presentamos 2 casos clínicos con diagnóstico mediante técnica de hibridación in situ (FISH) de inv dup(15) con afectación de la RCPWA, caracterizados por retraso mental, trastorno generalizado del desarrollo y fenotipo peculiar. El segundo caso clínico presentaba una epilepsia parcial compleja, con un electroencefalograma que ponía de manifiesto, principalmente, una actividad de fondo lentificada y paroxismos generalizados, caracterizados por complejos de puntas y polipuntas u ondas agudas/ondas. La respuesta a oxcarbazepina fue muy satisfactoria.
Conclusiones: Los CMS del cromosoma 15 que incluyen la RCPWA son casi siempre esporádicos y de origen materno. Causan una tetrasomía 15p y una tetrasomía parcial 15q. El objetivo del presente artículo es dirigir la atención de los pediatras a un síndrome neurogenético que puede sospecharse clínicamente, incluso en ausencia de confirmación citogenética.
Manejo y características de la fiebre de origen desconocido en pediatría
La fiebre de origen desconocido (FOD) es una entidad de difícil manejo si no se tienen en cuenta sus características clinicoepidemiológicas. El abordaje por parte del médico debe ser sistemático y ordenado para un correcto estudio; de no ser así, puede retrasarse o incluso obviar el diagnóstico de enfermedades potencialmente graves, así como comprometer futuros tratamientos. El objetivo de este trabajo es ofrecer al clínico una visión general de la FOD, así como contribuir a su mejor estudio mediante diversos protocolos de actuación. Se revisarán sus características, mostrando sus diferentes etiologías. Un aspecto básico es el diagnóstico; la historia clínica y la exploración física son fundamentales, pero el punto de mayor controversia son las pruebas de laboratorio y de imagen que se han de solicitar, aportando para ello un esquema sencillo de las pruebas que se requieren en cada momento.
Como conclusión, cabría destacar que la FOD no es una entidad infrecuente en pediatría, genera incomodidad en el médico y ansiedad en la familia, por lo que parece necesario un manejo adecuado del paciente y un uso racional de las pruebas complementarias para poder llegar a un diagnóstico correcto.
Neumonías adquiridas en la comunidad
Las neumonías adquiridas en la comunidad (NAC) y otras infecciones neumónicas son uno de los problemas de salud más importantes que afectan a los niños de todo el mundo. Hay 4-5 millones de muertes anuales registradas en niños menores de 5 años, de forma especial en los países en vías de desarrollo, donde figuran entre las más frecuentes causas de morbimortalidad. No es ése el caso de nuestro país, donde sin embargo constituyen una de las causas de mayor morbilidad, por lo que el problema que representan no es menor.
Quisiera agradecer a la Dirección de Acta Pediátrica Española su amabilidad por permitirnos exponer nuestra experiencia y una puesta al día de esta problemática infantil, cambiante en estos últimos años por la inmigración y el cambio epidemiológico, y quizás ecológico, que se está originado en nuestro medio por esta nueva situación.
Complicaciones de la dermatitis atópica severa
Sr. Director:
La dermatitis atópica (DA) es una enfermedad inflamatoria de la piel muy común en la infancia1.
Durante el primer año de vida se da en el 60% de los pacientes y durante los 5 primeros años en el 85%2. Su evolución a eritrodermia constituye un hallazgo excepcional3, 4.
Presentamos el caso de un recién nacido sano que nace por PEV, Apgar 9-10-10. Correctamente vacunado. La madre padece rinitis alérgica y tiene un hermano con DA. A los 15 días de vida presenta eritema facial, con empeoramiento progresivo a lo largo de los meses y afectación más intensa en flexuras.
Fue diagnosticado y tratado de DA con corticoides tópicos, antihistamínicos y emolientes.
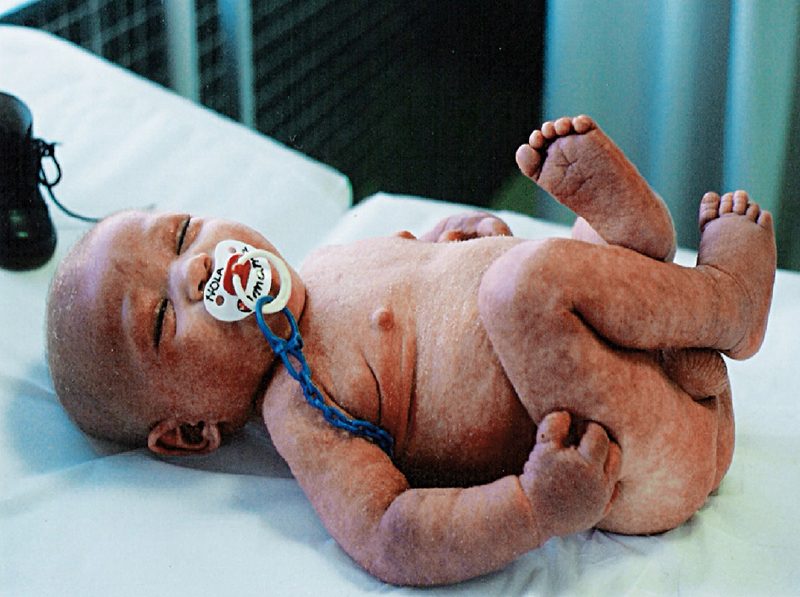
A los 8 meses evoluciona con agudización de su enfermedad, entra en eritrodermia y precisa ingreso (figuras 1 y 2).
Durante el ingreso se detecta sepsis por Staphylococcus aureus y secundariamente artritis de cadera.
Tras el tratamiento, se mantiene estable y al año de edad entra de nuevo en eritrodermia, siendo necesario tratarlo con corticoides por vía intravenosa (i.v.). Se detecta déficit proteico con hipoalbuminemia, por lo que precisa aporte de albúmina i.v. La eosinofilia se eleva hasta el 37,7% y la IgE a 5.390. Biopsia cutánea compatible con DA y biopsia ganglionar con adenitis reactiva. Las pruebas complementarias descartan histiocitosis, síndrome de Job y síndrome de Leiner5.
La DA es una enfermedad cutánea de comienzo en la infancia que ha aumentado mucho en la última década, fluctuando la prevalencia global actual entre un 10 y un 15% de la población6.
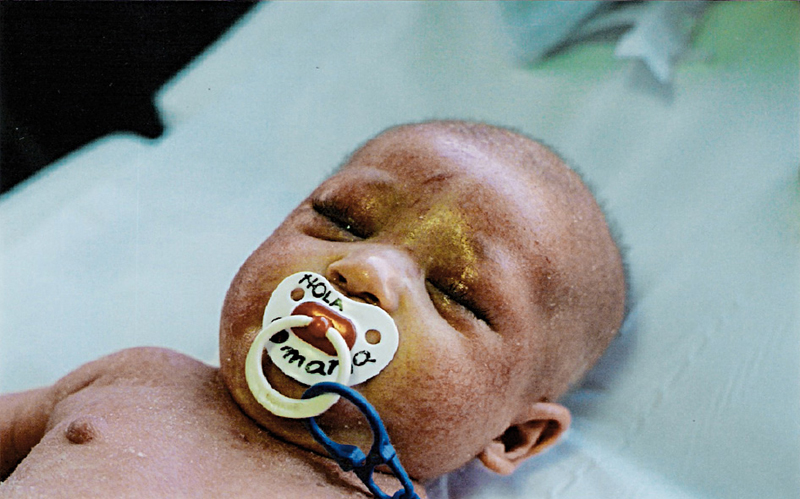
La edad de presentación de la enfermedad es, en el 60% de los casos, en el primer año de vida, pero es excepcional que aparezca antes de los 2 meses3,6. También es excepcional que la DA en fase de lactante tenga su aparición en flexuras (fase infantil)3, ya que en los lactantes suele aparecer en las superficies extensoras de las extremidades4.
La eritrodermia secundaria a DA es poco frecuente (ocurre en menos de un 1% de los pacientes)3. Muchas veces, como ocurre en el caso clínico descrito, su aparición está en relación con una sobreinfección.
Sus características clínicas son las de la DA, pero generalizada por todo el cuerpo. En general, es una eritrodermia descamativa con escama fina y fondo eritematoso, en la que llama la atención las numerosas lesiones de rascamiento secundarias al intenso prurito.
Es potencialmente mortal por insuficiencia cardiaca, con gasto elevado, infección sistémica por deterioro de la función barrera de la piel, pérdida de calor por vasodilatación cutánea, depleción proteica e inanición; por ello es de vital importancia un diagnóstico y tratamiento rápidos y correctos5.
Bibliografía
- Peramiquel Fonollosa L, Puig Sanz L. Dermatitis atópica. En: Fonseca Capdevila E, ed. Dermatología Pediátrica (VI). Madrid: Aula Médica 2005; 1.833-1.909.
- Vickers CFH. The natural history of atopic eczema. Acta Derm Venereol (Stockh) Suppl. 1980; 92: 113-115.
- Gerra Tapia A, Ruiz Rodríguez R, Ortiz Frutos JF. Dermatitis atópica. En: Foseca Capdevila E, ed. Dermatología Pediátrica (I). Madrid. Aula Médica 2000; 83-180.
- Lionel Fry, Charles N. Atlas de eccema atópico. Atlas Medical Publishing, Ltd. Edicion Española, 2004; 5-98.
- Ferrándiz Foraster. Eritrodermias infantiles. En: Fonseca Capdevila E, ed. Dermatología Pediátrica (I). Madrid: Aula Médica 2000; 15-79.
En Octubre de 1957 «Acta Pediátrica Española» publicaba...
CRÓNICA
«El niño hambriento debe ser alimentado», por el doctor Rodríguez Pedreira
ARTÍCULO REPRODUCIDO
«Las drogas tranquilizadoras y el escolar», por el doctor H. Freed
Circular del doctor Bosch-Marín a los puericultores del Estado
Neumotórax y neumomediastino espontáneos: presentación de un caso y revisión de la bibliografía
Tanto el neumomediastino como el neumotórax espontáneo son dos entidades poco frecuentes en la práctica clínica pediátrica. Difícilmente pensamos en ellas cuando se nos presenta un niño en urgencias con dificultad respiratoria y sin el antecedente de un traumatismo torácico, procedimiento quirúrgico o médico.
El neumomediastino se presenta con aire extraluminal dentro del espacio mediastínico, mientras que el neumotórax se presenta con aire en la cavidad pleural que provoca colapso pulmonar. Son espontáneos cuando ocurren sin antecedente de traumatismo o enfermedad pulmonar subyacente. Son más frecuentes en varones, y suelen acompañarse de enfisema subcutáneo secundario.
Debido a esta escasa frecuencia y a que los síntomas pueden llevar a interpretar diagnósticos erróneos o tardíos, el propósito de este artículo es tanto revisar las etiologías como la presentación clínica más frecuente de las dos entidades, así como evaluar las opciones de tratamiento oportunamente.
La comunicación interauricular: causa subyacente poco común de neumonía recurrente. Caso clínico y revisión de la bibliografía médica
Presentamos el caso de una lactante con infecciones respiratorias de repetición de las vías bajas y neumonías recurrentes (NR) en los 8 primeros meses de vida. Tras el estudio protocolizado inicial, no se encuentra una causa subyacente. Tras aparecer en una de las radiografías una dilatación de la aurícula derecha y el tronco pulmonar, se realiza una valoración cardiológica, incluida una eco-Doppler color, donde se aprecia una comunicación interauricular (CIA) grande, tipo ostium secundum, con repercusión hemodinámica. Las CIA pueden predisponer por diversos motivos (hiperaflujo, compresión extrínseca, aumento de secreciones, etc.) a la aparición de NR, aunque en algunas series extensas y en los protocolos diagnósticos se descartan a menudo las cardiopatías congénitas como causas subyacentes de NR.
Microcefalia, defecto de crecimiento y retraso mental. Dificultades diagnósticas para el síndrome alcohólico fetal
El síndrome alcohólico fetal (SAF) se define como un defecto congénito permanente causado por el consumo excesivo de alcohol materno durante el embarazo. Se caracteriza por un crecimiento disminuido, una alteración del sistema nervioso central y un conjunto de alteraciones faciales menores. La incidencia estimada es de 0,33-2,2/1.000 recién nacidos vivos en Estados Unidos. Hasta los años noventa no hubo una serie de criterios unificados y objetivos para llegar al diagnóstico de SAF. Se presenta el caso clínico de un niño de 5 años y 10 meses con este síndrome. El objetivo de este artículo es revisar los criterios diagnósticos del SAF y su actualización.
Infección congénita por citomegalovirus: valor de la resonancia magnética cerebral en el diagnóstico
Introducción: La infección congénita por citomegalovirus (CMV) es una afección frecuente en los países desarrollados. El objetivo de este trabajo es describir un caso clínico confirmado, cuya imagen por resonancia magnética (RM) cerebral revelaba anormalidades características de gran valor, que podrían incluso permitir un diagnóstico retrospectivo en caso de ausencia de sintomatología neonatal.
Caso clínico: Se trata de una recién nacida con microcefalia, ictericia y equimosis generalizadas, hepatomegalia, esplenomegalia y hallazgos analíticos compatibles con una infección congénita por CMV. Tras confirmarse dicha infección, se inicia tratamiento intravenoso con ganciclovir. Se realiza una RM, donde se aprecian lesiones multifocales en la sustancia blanca, con afectación predominante de las regiones profundas y presencia de las lesiones más importantes en las regiones parietal y temporal anterior. Se muestran y describen las imágenes de la RM de la paciente.
Discusión: La afectación de la sustancia blanca suele cursar con un patrón distintivo, según importantes fuentes bibliográficas. Las lesiones ubicadas en la sustancia blanca temporal anterior (incluidos los quistes en dicha localización) son particularmente sugestivas de una infección congénita por CMV. Nuestra paciente cumplía los criterios de RM característicos. Se realiza una revisión de la literatura médica, haciendo hincapié en los hallazgos de neuroimagen en la infección congénita por CMV.
Síndrome de esplenomegalia tropical en el diagnóstico diferencial de esplenomegalia masiva
Presentamos el caso de un niño de 11 años de edad, de origen guineano, enviado a nuestro hospital por presentar esplenomegalia masiva, anemia, eosinofilia y fiebre intermitente. El estudio etiológico inicial no fue concluyente. Finalmente, se estableció el diagnóstico de síndrome de esplenomegalia por hiperreactividad a la malaria, o síndrome de esplenomegalia tropical, para lo cual fueron esenciales la exclusión de otras causas y la respuesta al tratamiento antipalúdico; la biopsia hepática fue característica del cuadro.
Probióticos para el binomio madre-hijo (I)
El diseño de probióticos dirigidos a la población infantil ha despertado interés en los sectores médico e industrial, ya que podrían facilitar la creación de una barrera segura y eficaz frente a microrganismos patógenos y, además, contribuir a la maduración del tejido linfoide asociado a la mucosa intestinal. Actualmente, existen evidencias sobre el efecto beneficioso que algunos probióticos ejercen sobre la salud infantil, particularmente en lo concerniente a la reducción de la duración y severidad de los procesos diarreicos asociados a gastroenteritis infecciosas agudas, incluyendo aquellas asociadas a la antibioterapia. Por otra parte, ciertas cepas podrían ejercer efectos preventivos y terapéuticos sobre el eccema atópico y otras enfermedades de base alérgica, aunque serán necesarios más estudios clínicos antes de que se generalice el empleo de probióticos para el tratamiento de estas enfermedades.
Bebé colodión: manejo y proceso diagnóstico
El «bebé colodión» es una situación clínica poco frecuente que se presenta en el neonato y que es compartida por varias enfermedades y síndromes.
La alteración de la barrera epidérmica hace que el manejo y soporte del neonato sean fundamentales en los primeros días de vida, para luego llegar a un diagnóstico adecuado.
Presentamos un nuevo caso de esta entidad en la que la colaboración del neonatólogo y el dermatólogo es fundamental.
Diez casos de tiña incógnita en una consulta de pediatría. Un estudio retrospectivo (1985-2005)
El término «tiña incógnita» se utiliza para dermatofitosis con características clínicas no habituales, por utilización tópica de corticoides o inmunomoduladores.
Pacientes y métodos: Se exponen 10 casos diagnosticados durante 20 años, con una media de edad de 6,5 años y un tiempo de evolución medio de 2,7 meses.
Resultados: En 3 pacientes existía más de un caso familiar, lesiones micropapulosas, pustulosas y descamativas localizadas en la cara, el cuello o las extremidades, T. mentagrophytes en 7 casos y M. canis en uno.
Conclusiones: Las lesiones inusuales o difíciles de reconocer, tratadas previamente con corticoides o inmunomoduladores tópicos, requieren siempre un estudio micológico.
Prevalencia de la sensibilización a neumoalérgenos en nuestro medio
Los alérgenos son sustancias, habitualmente de naturaleza proteica, capaces de inducir en individuos predispuestos genéticamente (atópicos) anticuerpos IgE específicos, y que resultan inocuas para el resto de la población1-3.
Según la vía de entrada y el órgano de contacto, la respuesta inmunitaria o de hipersensibilidad alérgica dará lugar a manifestaciones clínicas oculares (conjuntivitis), digestivas (alergias alimentarias) o respiratorias (asma y/o rinitis).
Los alérgenos más frecuentemente relacionados con el asma son los que están presentes en el aire (aeroalérgenos o neumoalérgenos) y, al ser inhalados, ejercen sus efectos sobre la mucosa respiratoria, siendo los principales factores desencadenantes del asma y contribuyendo a la persistencia de síntomas en niños con asma establecida4.
Los principales neumoalérgenos sensibilizantes en nuestro medio, con una población bastante significativa, son los pólenes (gramíneas y olivo), los árboles ornamentales (arizónicas y plátano de sombra), el epitelio de animales, los ácaros del polvo y los hongos.
Protocolo de actuación en pacientes con displasia broncopulmonar/enfermedad pulmonar crónica (y II)
La displasia broncopulmonar/enfermedad pulmonar crónica de la infancia constituye un grupo heterogéneo de enfermedades respiratorias cada vez más frecuentes en nuestro medio debido, principalmente, a una mayor supervivencia de los recién nacidos de extremado bajo peso. Es una patología multisistémica muy compleja, con una etiopatogenia multifactorial, un cuadro clínico variado con participación de diferentes aparatos y sistemas, así como con muy diversas posibilidades diagnósticas y terapéuticas, que deberán conocerse en profundidad para establecer un buen control de esta enfermedad. Por ello, se requiere un seguimiento individualizado y un abordaje multidisciplinar, en el que es necesaria la implantación de un programa bien estructurado de intervención y seguimiento.
El objetivo de este trabajo es exponer los principales problemas asociados a los pacientes con displasia broncopulmonar/enfermedad pulmonar crónica de la infancia, así como elaborar un plan de actuación tras el alta del servicio de neonatología.
En Noviembre de 1958 «Acta Pediátrica Española» publicaba...
El doctor Vicente Giménez, de Valencia
ARTÍCULOS ORIGINALES
Trabajos doctrinales y casos clínicos
Electroforesis sobre papel y reacción del antígeno metílico en el Kala-azar infantil, por los doctores R. Marco Ahuir y G. Bellod García
Anemias infantiles en el litoral mediterráneo, por el doctor J. Selfa
Tres casos de meningitis hipertóxica (Waterhouse-Friderischen), por el doctor J.A. Ruiz-Santamaría
Anemia hemolítica grave en un lactante, curada por esplenectomía, por los doctores J. Rodrigo y V. Oliete
Embriopatías de origen rubeólico, por el doctor V. Milio
En Septiembre de 1957 «Acta Pediátrica Española» publicaba...
FIGURAS DE LA PEDIATRÍA
Doctor Carlos Sainz de los Terreros
ARTÍCULOS ORIGINALES
Trabajos doctrinales y casos clínicos
La celiaquía. Revisión y puesta al día, por el doctor Andrés González-Meneses Pardo
Estado actual de la cirugía del recién nacido, por el doctor Juan Garrido-Lestache Cabrera
La plantigrafía en la clínica pediátrica, por los doctores José María Mingo de Benito y Narciso Bermejo
Oxigenoterapia neonatal, por el doctor Joaquín Ramis Coris
Quince casos de tos ferina tratados con ACTH asociado a antibioterapia, por el doctor Dionisio Romero García-Pelayo
Trombopenia aguda posrubéola, por el doctor Jesús Martínez Fernández
Agenesia unilateral del nervio coclear
La agenesia del nervio coclear es un trastorno embriológico en la formación del oído interno que provoca una sordera neurosensorial. Presentamos el caso de un varón de 2,5 años de edad, que consulta por presentar retraso del lenguaje, hipoacusia del oído derecho y problemas del comportamiento. En la exploración se percibe una conexión adecuada con el medio, con un lenguaje limitado, y no se observa ningún hallazgo patológico en el examen neurológico.
Ante la sospecha de trastorno específico del lenguaje se solicitó un electroencefalograma, un estudio genético, una resonancia magnética (RM) y una valoración otorrinolaringológica (ORL) y psicopedagógica. La RM craneal mostró una agenesia del nervio vestibulococlear derecho, y el estudio ORL una hipoacusia neurosensorial derecha. Diagnosticado de una afectación grave del desarrollo del lenguaje receptivo-expresivo, no se ha podido valorar su relación con la pérdida auditiva total.
Las técnicas de imagen se han convertido en el método ideal para la detección de hallazgos patológicos en el oído interno, y deben realizarse ante toda pérdida de audición. Además, son útiles para la evaluación de los pacientes candidatos a la colocación de un implante coclear, ya que detectan anormalidades que pueden desaconsejar la intervención. Aunque las anomalías del conducto auditivo interno asociado con una agenesia del nervio coclear son, en principio, una contraindicación quirúrgica de implantación coclear, estos pacientes se podrían beneficiar de la estimulación eléctrica directa del núcleo coclear aplicando implantes en el tronco cerebral. En los niños con hipoacusia no sólo se altera el lenguaje, sino también las áreas emocional, social, cognitiva y motora, por lo que es necesario un tratamiento multidisciplinario.
Abdomen agudo como primera manifestación de crisis hemolítica
Presentamos el caso de un adolescente de 13 años, atendido en el servicio de pediatría de nuestro hospital, que consultó por presentar dolor abdominal y síntomas digestivos, y en cuya exploración y exámenes complementarios se detectaron signos de hemólisis. Tras las medidas de soporte iniciales, se realizó un estudio hematológico, en el que se detectó un déficit de la enzima glucosa-6-fosfato deshidrogenasa. Los familiares referían que el día anterior el paciente había ingerido habas por primera vez.
Nevo lipomatoso cutáneo superficial
Se presenta el caso de un niño afectado por un nevo lipomatoso cutáneo superficial localizado en el muslo. Se describen los aspectos epidemiológicos, anatomopatológicos y clínicos de esta entidad y se revisan los casos pediátricos publicados en la bibliografía científica de nuestro país.
La amnesia global transitoria: entidad infrecuente en la infancia
Introducción: La amnesia global transitoria (AGT) es un cuadro de presentación súbita caracterizado por una intensa amnesia anterógrada y una amnesia retrógada variable. La memoria inmediata está conservada y el resto de la exploración neurológica es normal. La recuperación es progresiva en pocas horas. Suele presentarse en pacientes adultos de mediana edad y en ancianos.
Caso clínico: Niña de 9 años de edad que presenta una amnesia anterógrada de inicio brusco, con memoria inmediata conservada y algún trastorno de la memoria retrógada, asociados a cefalea de intensidad leve. La exploración neurológica es normal. El episodio cede de forma espontánea en unas 4 horas. Posteriormente, la paciente presenta una cefalea leve y una amnesia del episodio. Se realiza una tomografía computarizada craneal, que es normal, por lo que el cuadro se orienta como AGT.
Discusión: La clínica descrita es característica de la AGT. La aparición de este cuadro suele ser espontánea, aunque puede existir un factor desencadenante. Se ha postulado que podría deberse a un fenómeno vascular amplio con isquemia transitoria del territorio de la vertebrobasilar, o a un fenómeno epiléptico o migrañoso. La migraña confusional aguda, típica de la infancia, tiene manifestaciones clínicas similares a las de la AGT. El diagnóstico de este proceso es clínico. Tiene un excelente pronóstico, con poca tendencia a la recidiva. Dada la presentación poco habitual en la edad pediátrica, en nuestro caso se realizó una prueba de imagen para descartar un accidente cerebrovascular. La clínica típica y la normalidad de las pruebas de imagen deben hacernos considerar este diagnóstico también en la edad pediátrica.
Síndrome de Gilbert: estudio de 12 observaciones y revisión de la bibliografía médica
El síndrome de Gilbert es el hallazgo de hiperbilirrubinemia indirecta leve-moderada con pruebas de función hepática normales y sin signos de hemólisis. Es un trastorno hereditario del metabolismo de la bilirrubina, benigno, con una prevalencia mundial cercana al 10%. Tiene un patrón de herencia variable, con polimorfismo genético. Se diagnostica mediante pruebas confirmatorias de provocación, como la prueba del ayuno, aunque el diagnóstico definitivo es genético. Su pronóstico es bueno y actualmente se discute sobre los diversos efectos de la hiperbilirrubinemia.
En este artículo, se revisan los casos diagnosticados de síndrome de Gilbert en el servicio de pediatría de un hospital universitario en los últimos años, y se describen las principales características halladas.
Fórmulas para lactantes sanos: principales novedades de la Directiva 2006/141/CE sobre preparados para lactantes y preparados de continuación
Objetivo: Esta revisión de la Directiva comunitaria de preparados para lactantes y de preparados de continuación analiza las modificaciones acaecidas tras la propuesta del Comité Científico para la Alimentación Humana de la Unión Europea (UE), la postura de la industria y la posición del Grupo Internacional de Expertos, liderado por la ESPGHAN, así como su plasmación normativa europea e internacional.
Metodología: Comparación de la Directiva 91/321/CEE (modificada por la Directiva 96/4/CE), el Informe de dicho Comité Científico del 2003 y las recomendaciones del Grupo Internacional de Expertos del 2005 con la Directiva 2006/141/CE y la Norma Codex 2007.
Conclusiones: La aprobación de estas dos nuevas normas ha permitido regular los avances científicos y tecnológicos sucedidos en los últimos 10 y 25 años, respectivamente, en temas de composición, etiquetado, presentación y publicidad de estos preparados. Sin embargo, quedan puntos controvertidos que serán probablemente objeto de futuras revisiones.
- vitaminas
- ácidos grasos
- energía
- minerales
- probióticos
- lípidos
- preparados para lactantes
- preparados de continuación
- proteínas
- aminoácidos
- nucleótidos
- hidratos de carbono
- FOS
- GOS
- introducción preparados de continuación
- declaraciones o alegaciones nutricionales
- declaraciones o alegaciones de propiedades saludables
- aplicación legal
- Volumen 65 número 8 septiembre 2007
Hamartoma fibroso de la infancia
El hamartoma fibroso de la infancia es un tumor raro, benigno, descrito por primera vez por Reye en 1956 con el término de «tumor fibromatoso subcutáneo de la infancia». Fue Enzinger, en 1965, quien definitivamente acuñó el término de «hamartoma fibroso de la infancia». Suele aparecer durante los primeros 2 años de vida, y es congénito hasta en un 15-20% de los casos. Es típica la presentación en forma de lesión única, asintomática, predominantemente en la región axilar, el tronco superior, la región inguinal o los genitales externos. La biopsia y la tinción con hematoxilina-eosina suelen ser suficientes para confirmar el diagnóstico, aunque las técnicas de inmunohistoquímica también pueden ser útiles. No hay evidencia de regresión espontánea del hamartoma fibroso de la infancia, pero tiene un pronóstico excelente, ya que se localiza exclusivamente en la dermis y el tejido celular subcutáneo, sin invadir los tejidos más profundos o las vísceras. El tratamiento de elección es la extirpación completa de la lesión, con tasas de recidiva muy bajas, incluso tras extirpaciones incompletas.
Morbilidad neonatal en el parto instrumentado: mención especial a la ventosa obstétrica
Introducción: El gran aumento en la utilización de diversos tipos de instrumentación obstétrica durante la extracción fetal, así como del número de cesáreas, es un tema muy controvertido en la actualidad. El objetivo de este trabajo es valorar la relación entre diferentes tipos de instrumentación y la incidencia de traumatismos obstétricos específicos.
Población y métodos: Estudio de casos y controles, realizado sobre una muestra de estudio de 103 recién nacidos, en los que como diagnóstico al alta figuró alguno de los relacionados con traumatismos perinatales: cefalohematoma, hemorragia subaponeurótica, subaracnoidea, epidural, subdural, parenquimatosa, fractura de clavícula y parálisis braquial.
Resultados: El vacuum fue el instrumento más utilizado, y constituyó un factor de riesgo respecto a la cesárea en la producción de cefalohematomas, hemorragias subgaleales, subaracnoideas, subdurales y fracturas de clavícula.
Discusión: El vacuum es, en números absolutos, el instrumento que se asocia a un mayor número de traumatismos. El alto porcentaje de traumatismos con el vacuum podría estar relacionado con el elevado número de inducciones que terminan en vacuextracción, lo que hace suponer que la modalidad de parto (espontáneo frente a inducido) es un factor esencial, asociado a la morbilidad del expulsivo; pero hay otros factores significativos además de las inducciones, como el tipo de campana usada durante la vacuextracción, el peso fetal, la anestesia epidural y la maniobra de Kristeller.
Síndromes de deficiencia de adhesión leucocitaria
Los síndromes de deficiencia de adhesión leucocitaria (leukocyte adhesion deficiency [LAD]) engloban un conjunto de patologías causadas por defectos en el reconocimiento, la adhesión y la migración de los leucocitos mieloides hacia los lugares de invasión microbiana, lo que provoca la falta de defensa innata del huésped frente a bacterias, hongos u otros microrganismos. Se distinguen dos tipos: LAD I y LAD II. Los síndromes LAD I y sus variantes están causados por mutaciones que impiden la expresión o la función de las integrinas de la clase -2 (integrinas CD11/CD18, o integrinas leucocitarias). Por el contrario, los sujetos con LAD II tienen unas características clínicas similares, pero conservan intacta la expresión y la función de las integrinas leucocitarias. El fundamento molecular de la deficiencia de tipo LAD II es un defecto en la glucosilación de los ligandos situados en los leucocitos, reconocidos por la familia de las moléculas de adhesión de las selectinas. Recientemente, se ha atribuido el defecto a mutaciones en un transportador de fucosa localizado en el aparato de Golgi. El establecimiento de las bases moleculares de los síndromes LAD ha generado nuevas perspectivas en el estudio de los mecanismos de acumulación leucocitaria, que son de gran relevancia en múltiples síndromes de inmunodeficiencias, así como en ciertas enfermedades inflamatorias que acaban desencadenando una lesión tisular.
Protocolo de actuación en pacientes con displasia broncopulmonar/enfermedad pulmonar crónica de la infancia (I)
La enfermedad pulmonar crónica de la infancia asociada a la displasia broncopulmonar constituye un grupo heterogéneo de enfermedades respiratorias cada vez más frecuentes en nuestro medio debido, principalmente, a una mayor supervivencia de los recién nacidos con un extremo bajo peso. Es una patología multisistémica muy compleja con una etiopatogenia multifactorial, una clínica variada, con participación de diferentes aparatos y sistemas, y muy diversas posibilidades diagnósticas y terapéuticas que deben conocerse en profundidad para establecer un buen control de la enfermedad. Por ello, se requiere un seguimiento individualizado y un abordaje multidisciplinar, que precisa la implantación de un programa bien estructurado de intervención y seguimiento.
El objetivo de este trabajo es exponer los principales problemas relacionados con los pacientes con enfermedad pulmonar crónica de la infancia/displasia broncopulmonar, así como elaborar un plan de actuación tras el alta del servicio de neonatología.
En Diciembre de 1958 «Acta Pediátrica Española» publicaba...
El doctor Luis Manchado, de Las Palmas
Trabajos doctrinales y casos clínicos
¿Qué pasa en el mundo con la mortalidad infantil?, por el doctor M. Blanco Otero
El niño y sus circunstancias, por el doctor J. de Cárdenas
«Los fines de semana», «los puentes» y «las excursiones en días festivos», por el doctor Pérez-Albert
Desnutrición maligna en el niño pequeño, por el doctor José Antonio de Paz Garnelo
En Junio de 1957 «Acta Pediátrica Española» publicaba...
NÚMERO DEDICADO A LAS JORNADAS MÉDICO-ESCOLARES
Advertencia
Crónica, por el doctor Sancho Martínez
Primer tema prefijado
Mesa: presidente, doctor Ruiz Santamaría (Valencia); secretarios, doctor García de Leániz (Madrid) y doctor Álvarez Suárez
(Gijón)
Primera parte
Profilaxis de la infección tuberculosa en la edad escolar, por el doctor López Morales
Segunda parte
La infección tuberculosa en la edad escolar a la luz de la moderna quimioterapia, por el doctor Monturiol
Aportaciones al tema
Dr. Sainz de los Terreros
Dr. Álvarez (P. Víctor)
Dr. Serrano Galnares
Dr. Tolosa Latour
Dr. Cruz Hernández
Dr. Rufilanchas
Dr. Cirajas
Dr. Saldaña
Dres. Lacaci González y Suescun Remón
Dr. Prandi Farrás
Dr. Santos Sanz
Dr. Fernández Crehuet
Dr. M. de la Iglesia
Resumen del presidente, doctor Ruiz Santamaría
En Julio de 1957 «Acta Pediátrica Española» publicaba...
NÚMERO DEDICADO A LAS JORNADAS MÉDICO-ESCOLARES
Advertencia
Crónica, por el doctor Sancho Martínez
SEGUNDO TEMA PREFIJADO
Mesa: presidente, doctor A. Muñoyerro Pretel (Madrid); secretarios, doctores Cirajas Labajo (Valladolid) y Rodríguez Gallego (Madrid)
PRIMERA PARTE
Alimentación y desarrollo del escolar español, por el profesor doctor Martínez García (Barcelona)
SEGUNDA PARTE
Desarrollo del escolar español, por el doctor Conde Gargollo (Madrid)
APORTACIONES AL TEMA:
Dr. Sainz de los Terreros
Dr. Serrano Galnares
Dr. Cirajas Labajo
Dr. Tolosa-Latour
Dr. P. Víctor Álvarez
Dr. Serigó Segarra
Dr. Santos Sanz
Dr. Ruiz Santamaría
Dr. Rufilanchas Salcedo
Dr. Prandi Farrás
Dres. García Gras, Escudero Tellechea y Gómez Jara
Resumen del presidente, doctor Muñoyerro Pretel
Tumor estromal gástrico de estirpe neural: ¿neoplasia aislada o primera manifestación del complejo de Carney?
Los tumores gastrointestinales de naturaleza estromal son especialmente infrecuentes en niños. Presentan de forma genérica 2 vías de diferenciación: hacia la estirpe muscular (variante mioide) o como célula neural. Carney publicó en 1978 el primer caso de un tumor de este tipo asociado a neoplasia pulmonar (condroma) y neural (paraganglioma extradrenal). Los 3 tumores presentan un origen ectodérmico común, lo que daría origen a la aparición sinérgica o secuencial de éstos a lo largo de la vida del individuo, y obligaría a realizar un estudio completo ante la aparición de cualquiera de ellos. Comunicamos el caso de una niña de 11 años que presentó un tumor estromal gástrico, cuya única manifestación clínica fue una anemia sintomática.
Anfotericina B liposomal y leishmaniasis visceral en un paciente inmunocompetente
Presentamos el caso de un lactante de 19 meses, previamente sano, con antecedentes de vivir en una zona semirrural y de tener un perro como animal de compañía, que presentaba la clínica de fiebre de 2 semanas de evolución y esplenomegalia gigante. La pancitopenia y la hipergammaglobulinemia características de la enfermedad estaban presentes.
En la biopsia de médula ósea no se detectó el parásito, pero la serología para Leishmania infantum resultó positiva. Se comenta la sensibilidad del aspirado de médula ósea en el diagnóstico.
El paciente recibió tratamiento con anfotericina B liposomal, y la respuesta fue excelente. Este caso amplía la poca experiencia acumulada respecto al tratamiento de esta enfermedad con anfotericina B liposomal, en particular en niños menores de 2 años.
Encefalocele adquirido en un paciente con enfermedad de Albers-Schönberg
La osteopetrosis es una rara enfermedad ósea caracterizada por una esclerosis del esqueleto y causada por un defecto en la resorción ósea por parte de los osteoclastos. La forma autosómica dominante de la osteopetrosis se divide en 2 subtipos. El tipo I implica un notable engrosamiento de la bóveda craneal. En el tipo II, o enfermedad de Albers-Schönberg, predomina la esclerosis vertebral y de la base del cráneo.
Las complicaciones más frecuentemente descritas de la osteopetrosis se localizan en el sistema nervioso, secundarias a la compresión de los pares craneales, los vasos sanguíneos y la médula espinal, por la oclusión gradual o una falta de desarrollo de los orificios craneales.
Se presenta el caso de un niño de 5 años, cuya enfermedad se inició con una proptosis ocular bilateral de aparición brusca, cefalea y vómitos, secundaria a un encefalocele producido por una fractura espontánea del techo de la órbita.
Recién nacida con nevo melanocítico congénito gigante «en calzón»
El nevo melanocítico congénito gigante es una lesión pigmentada de gran tamaño presente al nacimiento. Su incidencia es de 1/1.000-500.000 recién nacidos. La localización más frecuente es el tronco posterior, la cara, el cuero cabelludo y las extremidades. Puede tener una morfología curiosa «en bañador» o «capelina».
Se presenta el caso de una recién nacida con un nevo congénito gigante «en calzón», que ocupaba toda la zona genital, inguinal, glútea, superior de ambos muslos e inferior del tórax, con pigmentación color marrón oscuro y negro, e hipertricosis, así como 3 más pequeños en las zonas parietal izquierda, antebrazo derecho y mentón. Además, presentaba lesiones planas pigmentadas en las extremidades y el tórax. Las analíticas, las ecografías abdominal y cerebral, la radiografía de raquis óseo y la resonancia magnética medular eran normales. El resultado de la biopsia de piel afectada fue de nevo intradérmico.
La mayor complicación del nevo melanocítico congénito gigante, aparte del problema estético, es la malignización. Estos nevos pueden asociarse con melanosis neurocutánea hasta en un 25% de los casos. Es fundamental el inicio del tratamiento lo más precoz posible.
Proctocolitis hemorrágica en lactante exclusivamente alimentado al pecho
La lactancia materna es la forma ideal de alimentación del recién nacido. Entre sus múltiples ventajas está la de prevenir las enfermedades alérgicas. La colitis hemorrágica es una forma de intolerancia a las proteínas de leche de vaca, cuya frecuencia está aumentando especialmente en los niños lactados al pecho. Se debe pensar en ella en un lactante con hebras de sangre en heces sin afectación del estado general, en el que se ha descartado la existencia de una fisura anal y una infección gastrointestinal. El diagnóstico se basa fundamentalmente en la respuesta clínica a la dieta de exclusión. El diagnóstico de certeza raramente se obtiene, ya que el buen pronóstico y la buena evolución del cuadro hacen innecesarios los estudios invasivos. Sólo en casos de persistencia del sangrado se debe valorar la realización de una endoscopia con toma de biopsias. Su tratamiento es la dieta exenta de proteínas de leche de vaca. El tratamiento de los niños alimentados con fórmula es sencillo. Sin embargo, en los casos de lactancia materna exclusiva hay algunos interrogantes que permanecen sin aclarar.
Liquen «aureus»
El liquen aureus es una entidad rara, englobada dentro de un grupo de enfermedades denominadas dermatosis purpúricas pigmentarias. Todas ellas son, básicamente, capilaritis de origen desconocido, caracterizadas histológicamente por un infiltrado perivascular de linfocitos T, extravasación de eritrocitos y depósitos de hemosiderina. Aparece predominantemente en niños y adultos jóvenes de cualquier raza, con más frecuencia en hombres que en mujeres. La biopsia cutánea confirma el diagnóstico. Las lesiones cutáneas suelen ser asintomáticas, aunque en algunos casos puede aparecer un prurito intenso. Tienden a la cronicidad, y se ha descrito, sobre todo en niños, la resolución espontánea al cabo de varios años. La respuesta al tratamiento suele ser bastante limitada. Los corticoides tópicos potentes rara vez son eficaces; en algunos casos, se ha intentado el tratamiento con fototerapia (PUVA), pimecrolimus o pentoxifilina, con resultados variables.
Distrofia de las veinte uñas y dermatitis atópica en un niño de 8 años
La distrofia de las veinte uñas es una entidad poco frecuente que se puede asociar con diversas dermatosis, como la alopecia areata, el liquen plano o la dermatitis atópica. Suele afectar a varones de 10-20 años. Las uñas de estos pacientes se muestran deslustradas y presentan una estriación lineal. Su diagnóstico es clínico, si bien los casos dudosos pueden precisar la realización de una biopsia ungueal. En cuanto al tratamiento, se han descrito resultados aceptables con la administración de corticoides por distintas vías, como la intralesional, la intramuscular y la oral. En este artículo se presenta un caso de distrofia de las veinte uñas secundaria a dermatitis atópica en un paciente de 8 años.
Síndrome de «shock» tóxico: a propósito de dos casos
Los síndromes de shock tóxico y shock similar a tóxico son entidades causadas por superantígenos que ponen en marcha una respuesta sistémica que determina sus características clínicas. Exponemos dos casos detectados en nuestro hospital que cumplen los criterios diagnósticos de síndrome de shock tóxico al presentar una temperatura ≥38,9 ºC, un exantema con posterior descamación, una hipotensión arterial y una afectación de 3 o más órganos. El primero evolucionó hacia un fallo multiorgánico con cultivos negativos, por lo que se diagnosticó de etiología probablemente estafilocócica. El paciente experimentó un deterioro progresivo, sin respuesta a las medidas de soporte, y falleció. El segundo caso se manifestó como una fascitis necrosante de la pared abdominal, que precisó un desbridamiento quirúrgico. El crecimiento de Streptococcus pyogenes se detectó en la faringe y en el exudado de la herida quirúrgica. Su evolución fue favorable, por lo que recibió el alta con una estenosis traqueal como única secuela.
Los dos casos demuestran la importancia de la sospecha clínica en estos cuadros para pautar el tratamiento antibiótico e instaurar una terapia de soporte con rapidez.
Síndrome de escaldadura estafilocócica de presentación neonatal
Introducción: La incidencia del síndrome de escaldadura estafilocócica en neonatos parece haber aumentado en los últimos años.
Métodos: Revisamos los casos de síndrome de escaldadura estafilocócica de presentación neonatal, controlados en nuestro servicio en los últimos 10 años (1997-2006).
Resultados: Durante este periodo, se diagnosticaron 26 casos de síndrome de escaldadura estafilocócica, 4 de ellos (15%) en el primer mes de vida. Todos presentaron eritrodermia, ampollas, costras perinasales, fisuración perioral y conjuntivitis purulenta. En ningún caso apareció fiebre ni elevación de reactantes de fase aguda. En todos los pacientes se aisló Staphylococcus aureus sensible a meticilina en el frotis nasal y conjuntival. Se realizó un tratamiento con cloxacilina intravenosa durante una media de 7 días, y los pacientes presentaron descamación en láminas sin lesiones residuales.
Conclusiones: El síndrome de escaldadura estafilocócica se presenta en el periodo neonatal hasta en el 15% de los casos. El tratamiento temprano con cloxacilina intravenosa permite una evolución favorable, e iguala el buen pronóstico característico del resto de edades.
Morbilidad neonatal en el parto instrumentado: mención especial a la ventosa obstétrica
El aumento de las lesiones en el periparto, asociadas al cada vez más creciente uso de instrumentación, ha llevado a plantear en numerosas ocasiones intensos debates sobre las indicaciones y la problemática derivada de su cada vez mayor indicación en la práctica obstétrica diaria. El vacuum es un instrumento bastante seguro, de modo que las lesiones pueden existir, pero son mucho menos frecuentes con una correcta aplicación, y la gravedad de las mismas se ve influida no sólo por la instrumentación, sino también por una serie de factores que hay que tener en cuenta en el periparto, siendo uno de los principales el desarrollo de la vacuextracción. Entre la lesiones que nos encontramos asociadas al uso del vacuum aparecen desde algunas de elevada incidencia pero de escasa significación clínica como el cefalohematoma o el hematoma subgaleal, hasta otras de escasa frecuencia pero de extrema gravedad y riesgo vital para el recién nacido, como el hematoma epidural o el subdural.
Reducción de la frecuencia de la cetoacidosis diabética en niños de 0-14 años
Sr. Director:
La diabetes mellitus insulinodependiente (DMID) es la enfermedad crónica infantil y la endocrinopatía pediátrica más frecuente. Hay una gran variación mundial en su incidencia por razones aún desconocidas, aunque influyen factores geográficos, raciales, ambientales y de predisposición genética de las diversas poblaciones. La DMID, o tipo 1, se presenta en un 10-30% de los niños en forma de cetoacidosis diabética (CAD), con una mortalidad del 1-10%, y en los últimos años se ha observado un claro descenso de la CAD como forma de inicio1.
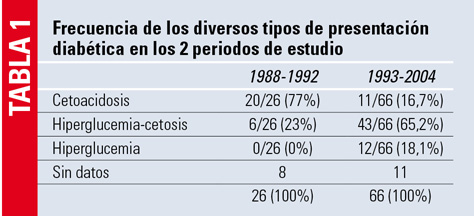 En el año 1994 publicamos en esta revista los datos de la incidencia de la DM tipo 1 en nuestra área en la población pediátrica de 0-14 años entre los años 1998 y 19922. En dicho trabajo aportamos la frecuencia de los diversos tipos de presentación diabética (cetoacidosis, hiperglucemia-cetosis e hiperglucemia). Desde entonces, han pasado 12 años, y con la misma metodología hemos observado, entre los años 1993 y 2004, un notable descenso en la CAD (–60,3%) y un correspondiente aumento de las formas más leves de presentación (tabla 1). En otras series españolas, la frecuencia de cetoacidosis como forma de presentación de la dibaetes es mayor (31,7% en Galicia y 24,9% en Málaga)3,4 que en la nuestra (16,7%).
En el año 1994 publicamos en esta revista los datos de la incidencia de la DM tipo 1 en nuestra área en la población pediátrica de 0-14 años entre los años 1998 y 19922. En dicho trabajo aportamos la frecuencia de los diversos tipos de presentación diabética (cetoacidosis, hiperglucemia-cetosis e hiperglucemia). Desde entonces, han pasado 12 años, y con la misma metodología hemos observado, entre los años 1993 y 2004, un notable descenso en la CAD (–60,3%) y un correspondiente aumento de las formas más leves de presentación (tabla 1). En otras series españolas, la frecuencia de cetoacidosis como forma de presentación de la dibaetes es mayor (31,7% en Galicia y 24,9% en Málaga)3,4 que en la nuestra (16,7%).
Sin poder ofrecer una explicación exacta de esta notable reducción, creemos que, entre otros factores, pudo influir nuestra insistencia en el diagnóstico precoz mediante una tira reactiva de orina a todos los MIR de pediatría y de medicina de familia que han pasado por nuestro servicio. Con esta información no se debe demorar más tiempo la remisión al hospital de los casos con un cuadro clínico sugestivo de esta enfermedad.
El mejor estado en el que los niños han llegado a nuestro hospital ha evitado numerosos ingresos en la unidad de cuidados intensivos y ha reducido el sufrimiento de los pacientes, de sus familias y de los médicos responsables de su tratamiento.
Bibliografía
- Molina Font JA. Tratamiento de la cetoacidosis diabética. En: Pombo M, ed. Tratado de endocrinología pediátrica, 2.ª ed. Madrid: Ediciones Díaz de Santos, 1997; 1.101-1.111.
- Aleixandre FA. Incidencia de la diabetes mellitus tipo I en población infantil de 0 a 14 años (1988-1992). Acta Pediatr Esp. 1994; 52: 155-160.
- Cepedano Dans A, Barreiro Conde J, Pombo Arias M, y Grupo de Diabetes Infantil de Galicia. Incidencia y características clínicas al manifestarse la DMI en niños de Galicia (España, 2001-2002). An Esp Pediatr. 2005; 62: 123-127.
- López Siguero JP, Lora Espinosa A, Martínez MJ, Martínez Valverde A. Incidencia de DMID en niños (0-14 años) en Málaga, 1982-1988. An Esp Pediatr. 1992; 37: 485-488.
Síndrome de activación de macrófagos y artritis juvenil de inicio sistémico
El síndrome de activación de macrófagos es una activación anómala del sistema histiocitario e indica una respuesta inadecuada del sistema inmunitario. Puede ser primario o reactivo; en los casos secundarios se asocia sobre todo con enfermedades de origen autoinmunitario, especialmente la artritis idiopática juvenil de inicio sistémico (AIJS). Su etiología es aún desconocida, pero se han implicado diferentes agentes infecciosos. Se define como un cuadro clínico agudo y grave de insuficiencia hepática, coagulopatía de consumo y encefalopatía, asociado con la presencia inconstante en la médula ósea de macrófagos activados con signos de hemofagocitosis. El diagnóstico y el tratamiento precoz mejoran el pronóstico de esta entidad potencialmente mortal.
Presentamos el caso de una niña de 18 meses que había sido diagnosticada de AIJS 9 meses antes y que, tras una infección por varicela, desarrolló un cuadro de fiebre alta, con un empeoramiento progresivo de su estado general hasta desarrollar un fracaso multisistémico, y en la que los datos de la autopsia revelaron signos de hemofagocitosis.
Edema angioneurótico hereditario en pediatría. Consideraciones a propósito de 3 casos
El edema angioneurótico hereditario (EAH) es una enfermedad sistémica poco frecuente, autosómica dominante y caracterizada por ataques recurrentes de edema localizado del tejido subcutáneo de la piel o las mucosas del tracto gastrointestinal y respiratorio alto.
Se origina por la ausencia o la deficiencia funcional del inhibidor de la C1 esterasa (C1-INH), que en condiciones normales inhibe la actividad proteolítica de la primera fracción del complemento (C1). Ello origina la activación incontrolada de las cascadas del complemento, la coagulación, la fibrinólisis y las cininas, con la consiguiente producción de moléculas vasoactivas responsables de las crisis de angioedema.
Se presentan 3 casos de angioedema hereditario en pacientes pediátricos, de los que se revisan los aspectos clínicos y terapéuticos más relevantes de la entidad.
Síndrome de «cri du chat». Comunicación de un nuevo caso y revisión
El síndrome de cri du chat, o monosomía 5p-, es una rara anomalía genética debida a la deleción de un segmento del brazo corto del cromosoma 5, que muestra una gran variabilidad fenotípica y citogenética. Su incidencia varía de 1/15.000 a 1/50.000 recién nacidos vivos, aunque es posible que su frecuencia sea mayor. Se comunica un nuevo caso neonatal con algunas de las características fenotípicas del síndrome, con una deleción en 5p15.2 por translocación materna, que precisó técnicas moleculares para su confirmación; asimismo, se revisan los aspectos clínicos y citogenéticos más interesantes de esta afección.
Fallo cardiaco en un neonato: hemangioma hepático, manejo y revisión de la bibliografía médica
Los hemangiomas constituyen los tumores hepáticos vasculares más frecuentes durante la infancia. Casi siempre son de naturaleza benigna, y la mayoría de ellos se diagnostica casualmente, sin presentar relevancia clínica. Cuando son sintomáticos, se recomienda inicialmente realizar tratamiento médico con corticoides. Cuando falla el tratamiento médico en el control de los hemangiomas sintomáticos, deben aplicarse medidas más agresivas, como la ligadura de la arteria hepática, la embolización, la resección hepática, etc. Describimos el caso de un hemangioma hepático neonatal resistente al tratamiento médico, en el que se realizó exitosamente una ligadura selectiva de la arteria hepática.
El método de la madre canguro
Desde hace tiempo, las unidades de neonatología están tratando de introducir los cuidados centrados en el desarrollo, que pretenden mejorar el desarrollo del prematuro a través de intervenciones especiales, entendiendo al recién nacido y a su familia como una unidad. En estos cuidados se incluye el método de la madre canguro (MMC), que se define como el contacto piel con piel entre la madre y el niño prematuro de la forma más precoz, continua y prolongada posible con lactancia materna, para que ambos se beneficien de sus ventajas. El MMC debería ofrecerse a todos los niños prematuros o recién nacidos a término enfermos como una alternativa al cuidado en la incubadora, dado que es efectivo para el control de la temperatura, reduce el riesgo de infecciones y de episodios de apnea, favorece la lactancia materna, permite y fortalece la vinculación madre/padre-hijo, devuelve a los padres el protagonismo del cuidado de sus hijos y reduce la estancia hospitalaria.
Mastocitosis cutánea
Definimos mastocitosis como un conjunto de trastornos clínicos, producidos todos ellos por una proliferación de mastocitos y una acumulación en varios órganos, entre los cuales el más común es la piel. La mastocitosis cutánea fue descrita por primera vez por Nettleship y Tay, en 1869, quienes asumieron que la acumulación de mastocitos quedaba limitada únicamente a la piel. Y no es hasta 1949 cuando Ellis describió por primera vez una forma sistémica de mastocitosis. Aproximadamente, un 80% de los pacientes con mastocitosis presenta sólo una afectación cutánea, mientras que el 20% restante tiene una mastocitosis sistémica, y en más de la mitad de éstos aparecen también lesiones cutáneas. El diagnóstico se basa fundamentalmente en los hallazgos clínicos e histológicos. En general, el pronóstico en la edad pediátrica es bueno, con tendencia a la resolución espontánea antes de la pubertad. Aun así, un 15-30% de los niños en quienes la enfermedad persiste durante la edad adulta desarrollará una afectación sistémica.
Nutrición, actividad física y masa ósea en el adolescente
La adolescencia es una etapa crítica en el establecimiento de la salud ósea, que representa la oportunidad de influir en los factores que determinen la magnitud de las ganancias óseas. Hay una serie de factores ambientales importantes que cabe tener en cuenta, como la ingesta de calcio y la actividad física habitual, que interactúan entre ellos. Parece que el esqueleto responde mejor a los complementos de calcio en las primeras etapas de la vida, antes del inicio de la pubertad; después del desarrollo puberal los efectos no son tan importantes. Sin embargo, diferentes autores subrayan que la administración de un complemento de calcio que no vaya acompañado de unos hábitos físicamente activos y/o de la práctica de ejercicio físico no tiene repercusiones relevantes sobre la masa ósea durante la adolescencia.
Cefalea en la infancia y la adolescencia. Aportaciones basadas en la evidencia
La valoración de la cefalea en la infancia no ha adquirido hasta fechas muy recientes la importancia que tiene desde el punto de vista epidemiológico, y es el problema neurológico más importante dentro de la patología atendida. Ello se debe a que el dolor de cabeza se asocia con procesos patológicos muy comunes y frecuentes en la edad pediátrica. Se estima que aproximadamente el 5% de los niños padece o ha padecido alguna vez un dolor de cabeza. Al igual que en la edad adulta, en la infancia la mayoría de las cefaleas son primarias. Una historia bien realizada y una exploración general y neurológica completa son la clave para un diagnóstico correcto. Sin embargo, hasta en un 30% de los casos no se puede establecer la causa del dolor de cabeza. Actualmente, la mayoría de los autores acepta los criterios de la Sociedad Internacional de Cefalea por su especificidad. En algunos casos, sobre todo ante la presencia de síntomas o signos de alarma, tendremos que completar el examen mediante la realización de exploraciones complementarias para descartar que se trate de una cefalea secundaria.
Endocarditis neumocócica: revisión a propósito de un caso y nueva pauta de tratamiento asociada a fosfomicina
Se presenta un interesante caso de endocarditis bacteriana en un varón de 12 años de edad, que asociaba como patología de base una comunicación interventricular membranosa, que presentó buena respuesta al tratamiento asociado con penicilina en altas dosis, gentamicina y fosfomicina.
Se adjunta una revisión bibliográfica acerca de los distintos tratamientos antibióticos descritos, incluidas las pautas menos habituales. Se incluye en la discusión una extensa explicación de la asociación al tratamiento antibiótico de fosfomicina.
En Mayo de 1957 «Acta Pediátrica Española» publicaba...
FIGURAS DE LA PEDIATRÍA
El doctor Loste Echeto, de Huesca
ARTÍCULOS ORIGINALES
Trabajos doctrinales y casos clínicos
Evolución de la cirugía cardiovascular en el último cuarto de siglo, por el doctor Pérez de Petinto
Estado actual de las cardiopatías congénitas, por el doctor Muñoz Calero
Conducta quirúrgica a seguir en la hipertrofia congénita de píloro, por el doctor Garrido-Lestache Díaz
Hernia estrangulada en el recién nacido, por el doctor García Andrade
Tratamiento quirúrgico de las anomalías anorrectales, por el doctor Juan G. Lestache Cabrera
Problemas estomatológicos del recién nacido, por el doctor Pedro García Gras
Oportunidad de intervención del labio leporino, por el doctor B. Vilar-Sancho Altet
Adenitis cervical por «Mycobacterium bovis»
Mycobacterium bovis es un microrganismo que origina fundamentalmente tuberculosis en el ganado. Pertenece a la familia del complejo Mycobacterium tuberculosis y es responsable de un porcentaje escaso de todas las tuberculosis en humanos. La incidencia es mayor en países en vías de desarrollo donde no hay un control sociosanitario de la tuberculosis del ganado. La vía de contagio suele ser digestiva por la ingesta de leche infectada no pasteurizada, por lo que las manifestaciones más frecuentes son extrapulmonares, entre las que destacan la linfadenitis cervical y la tuberculosis digestiva. El tratamiento debe hacerse con fármacos tuberculostáticos de primera línea, teniendo en cuenta que este microrganismo posee una resistencia intrínseca a la pirazinamida.
En nuestro medio debemos sospechar esta etiología ante un caso de tuberculosis en un paciente procedente de una zona donde la tuberculosis en el ganado no esté controlada.
Presentamos el caso de una niña de 11 años de origen marroquí, con tuberculosis ganglionar cervical debida a M. bovis.
Mucopolisacaridosis tipo I: evolución clínica de 2 pacientes tras 30 meses de tratamiento enzimático sustitutivo
La mucopolisacaridosis tipo I (MPS I) es una enfermedad lisosomal hereditaria producida por un déficit enzimático de a-L-iduronidasa, que da lugar a una acumulación de los glucosaminglucanos (GAG) dermatán y heparán sulfato en los órganos y los tejidos, así como a un aumento de su excreción urinaria. Hay tres subtipos: la enfermedad de Hurler (MPS IH) es la más grave, la enfermedad de Scheie (MPS IS) es la más leve y la enfermedad de Hurler-Scheie (MPS IHS) es la forma intermedia.
Es una enfermedad crónica y progresiva, con manifestaciones multisistémicas. La disminución de la capacidad pulmonar y los síntomas de obstrucción de las vías altas (síndrome apnea-hipopnea del sueño), la afectación cardiovascular y los problemas articulares son los que causan mayor morbimortalidad.
El trasplante de médula ósea o de células madre hematopoyéticas, junto con el tratamiento enzimático sustitutivo, constituyen los principales pilares del tratamiento.
Presentamos 2 casos de MPS I que han recibido tratamiento sustitutivo con enzima recombinante a-L-iduronidasa durante 30 meses y un tercer caso de un paciente que lo ha iniciado hace 3 meses. Ninguno ha presentado complicaciones atribuibles al tratamiento.
Protrusión perineal infantil
La protrusión perineal infantil es una entidad descrita recientemente, con una morfología de las lesiones, localización y prevalencia características, sobre todo en las niñas prepuberales. Presentamos un nuevo caso de esta infrecuente entidad.
Tumoraciones cervicales
Las tumoraciones cervicales son un proceso muy frecuente en la consulta de pediatría. Se deben valorar el momento de aparición, localización y tamaño para suponer su origen. En caso de tumoración que no mejora con antibióticos, en triángulo posterior o supraclavicular, fija o mayor de 2,5 cm, conviene que sea valorada por un cirujano para descartar la patología maligna.








