
Agenesia renal y orquiepididimitis: ¿en qué debemos pensar?
Resumen
El síndrome de Zinner es una alteración en el desarrollo embriológico poco común, que consiste en la asociación de una dilatación quística de la vesícula seminal con agenesia renal ipsilateral.
Presentamos el caso de un varón de 14 años diagnosticado de síndrome de Zinner a raíz de un cuadro de orquiepididimitis izquierda y hematuria. Entre los antecedentes personales del paciente destaca una agenesia renal izquierda con función renal conservada. Debido a la sospecha clínica se realizó una resonancia magnética (RM), en la que se objetivó uréter izquierdo dilatado y tortuoso, confirmándose su desembocadura en vesícula seminal izquierda.
Clínicamente suele manifestarse con episodios recidivantes de epididimitis, trastornos miccionales o alteraciones eyaculatorias, pudiendo aparecer a cualquier edad, aunque lo más frecuente es durante la segunda y tercera décadas de la vida.
La RM es la técnica diagnóstica de elección. El tratamiento depende directamente de la clínica. La cirugía suele reservarse para los pacientes sintomáticos o para aquellos quistes que se detectan a una edad temprana, para evitar posibles complicaciones.
Hematuria recidivante como síntoma guía del síndrome del desfiladero aortomesentérico
Resumen
Introducción: Este síndrome define la compresión de la vena renal izquierda entre la aorta y la arteria mesentérica superior, provocando una presión elevada de dicha vena renal con posible desarrollo de venas colaterales. Clínicamente, aparece hematuria intermitente, proteinuria con o sin dolor abdominal o en hemiabdomen izquierdo.
Casos clínicos: Presentamos 15 niños de entre 7 y 13 años, la mayoría con examen físico normal y clínica de dolor abdominal o del flanco izquierdo con presión arterial normal (46,6%); hallazgos urinarios: hematuria no glomerular (20%) y proteinuria de rango no nefrótico (20%), con microalbúmina inferior a 300 mg en orina de 24 horas. Un paciente (6,7%) presentó proteinuria asociada a glucosuria e hipertensión.
Las mediciones de ultrasonografía Doppler del diámetro anteroposterior (AP) y las velocidades máximas de la vena renal izquierda son diagnósticas: dilatación y enlentecimiento del flujo proximal a la pinza aortomesentérica, disminución del calibre con flujo acelerado a nivel distal y ángulo de la pinza menor de 30º. En dos casos precisó angio-TAC, mostrando la vena renal izquierda comprimida entre la aorta y la arteria mesentérica superior. La angiografía por RM ofrece una excelente definición anatómica. El tratamiento es conservador. Puede ser necesario tratamiento con inhibidores de la enzima convertidora de la angiotensina, intervenciones quirúrgicas o procedimientos extravasculares.
Conclusión: Sospechar SHVR ante la presencia de hematuria, proteinuria, dolor abdominal recurrente, diagnóstico que requiere alto índice de sospecha. Precisa ecografía. En casos seleccionados, angio-TAC, RM o flebografía, siendo esta última el gold standard para el diagnóstico, que no suele precisarse.
Orina litogénica e infección urinaria por bacteria poco habitual en una mujer adolescente
Introducción: Hay diversos factores predisponentes en la vía urinaria para la presencia de bacterias, entre ellos una orina litogénica, como puede ser la hipocitraturia.
Los Corynebacterium spp. se describen actualmente asociados a infección en relación con la manipulación instrumental de la vía urinaria, como el C. amycolatum.
Caso clínico: Adolescente de 13 años con clínica de cólico renal, que precisa ingreso para control del dolor. El cólico está en el contexto de antecedentes familiares de litiasis cálcica; en la orina se obtuvo el aislamiento monomicrobiano de Corynebacterium aurimucosum, en dos muestras de orina repetidas. Sólo con tratamiento antibiótico adecuado según antibiograma, con amoxicilina-clavulánico, desaparecieron el dolor cólico y la disuria.
Conclusión: Corynebacterium aurimucosum es una bacteria propia de la flora saprofita del aparato urogenital femenino, en nuestro caso está asociado a la clínica de cólico renal e infección urinaria. En determinadas situaciones, algunos gérmenes poco virulentos e incluso comensales urogenitales, podrían comportarse como patógenos. Ello subraya la importancia de la recogida de un urocultivo previo al inicio de la antibioterapia empírica, al menos en determinadas circunstancias, como la orina prelitiásica.
Cólico renal como forma de expresión de adenoma paratiroideo
El hiperparatiroidismo primario es una entidad muy poco frecuente en la edad pediátrica. La mayoría de los casos son esporádicos y debidos a adenomas. Debe sospecharse ante una hipercalcemia con parathormona elevada. La resección quirúrgica es el tratamiento de elección, con una evolución favorable de los casos en general.
Presentamos el caso de una niña de 9 años de edad con adenoma paratiroideo, iniciado en forma de cólico renal e hipercalcemia. La gammagrafía tiroidea con tecnecio-99 sestamibi confirmó el diagnóstico. Tras la intervención quirúrgica presentó una buena evolución, con normalización de las concentraciones de calcio.
En el diagnóstico diferencial del cólico renal en niños, hay que tener en cuenta la posibilidad subyacente de enfermedades raras con tratamiento específico, como el adenoma paratiroideo.
Transfusión en anemia hemolítica autoinmune: una cuestión vital. A propósito de un caso
Introducción: La anemia hemolítica autoinmune (AHAI) es rara en la infancia, con una incidencia anual estimada de 1 cada 80.000 personas en la población general. La anemia suele ser moderada y bien tolerada por el paciente. Las transfusiones de concentrados de hematíes no están indicadas habitualmente, salvo en situaciones de riesgo vital.
Caso clínico: Se presenta el caso de una paciente con AHAI por anticuerpos IgG calientes. Tras el diagnóstico se inició tratamiento corticoideo, y la paciente requirió una transfusión de concentrados de hematíes en 2 ocasiones y 3 dosis de inmunoglobulinas como tratamiento adyuvante.
Conclusiones: Por regla general debe evitarse la transfusión de concentrados de hematíes en la AHAI, salvo en situaciones que comprometan la vida del paciente, puesto que podrían intensificar la hemolisis. Sin embargo, en algunos de estos pacientes la enfermedad puede presentarse como una emergencia que aconseje la transfusión inmediata pese a los riesgos que ello implica, ante la presencia de signos de hipoxia cerebral, cardiaca o renal. Se debe individualizar el caso de cada paciente, y valorar más que nunca el riesgo-beneficio.
Síndrome de piel escaldada neonatal producido por estafilococo resistente a la meticilina
El síndrome de piel escaldada estafilocócica es una enfermedad cutánea ampollosa poco frecuente, causada por una toxina exfoliativa. La mayoría de los casos ocurren en menores de 5 años, con una mortalidad entre el 4 y el 11%. Los neonatos tienen una susceptibilidad aumentada, ya que a la incompetencia inmunológica se añade la inmadurez de la función renal, que limita el aclaramiento de las toxinas. Los casos debidos a estafilococo resistente a la meticilina son poco frecuentes en esta población, y un diagnóstico y tratamiento precoz son fundamentales para disminuir la morbimortalidad. Presentamos el caso de un neonato de 5 días de vida con un síndrome de piel escaldada estafilocócica, producido por un estafilococo resistente a la meticilina, que presentó buena evolución con el tratamiento.
Advanced ultrasound techniques for pediatric imaging / Estado general de salud y satisfacción vital en niños con enfermedades crónicas
Advanced ultrasound techniques for pediatric imaging
Hwang M, Piskunowicz M, Darge K. Pediatrics. 2019; 143: e20182609.
Resumen
La ecografía constituye una herramienta esencial en el abordaje diagnóstico en pediatría, por su disponibilidad, seguridad y bajo precio. En esta revisión se presentan los avances más recientes en la técnica ecográfica y su aplicabilidad clínica, que se concretan en mejoría de la sensibilidad de la escala de grises convencional, el uso de Doppler color y la posibilidad de obtener una información funcional adicional. Además, la revisión ilustra el empleo de cada técnica en casos clínicos paradigmáticos.
Ecografía de contraste
En este tipo de ecografía se usan agentes de contraste que son microburbujas de gas microencapsuladas con fosfolípidos de 2-3 µm de tamaño, que generan una señal potente de ultrasonidos. La técnica requiere la inyección del contraste en una vía periférica, que se elimina por exhalación a través de los pulmones, por lo que es más seguro que el contraste usado en la tomografía computarizada (TC) o en la resonancia magnética (RM). Además, elimina la necesidad de sedación en niños.
La principal utilidad de esta prueba en niños pequeños es su capacidad para diagnosticar algunas lesiones focales benignas en el hígado, como los hemangiomas, la hiperplasia nodular o la infiltración grasa. Al ser capaz de delimitar bien el patrón de vascularización de una lesión, puede ser de ayuda para distinguir entre lesiones benignas y malignas. En las lesiones malignas, generalmente muy vascularizadas, el fenómeno de aclarado rápido del contraste es uno de los rasgos más característicos de la ecografía de contraste y, además, puede identificar áreas de necrosis frente a áreas de tumor viable, por lo que sirve de guía para las biopsias dirigidas. Como es útil para el seguimiento de estas lesiones, disminuye la necesidad de realizar TC o RM.
También es útil para el diagnóstico de la isquemia o el daño en un órgano (p. ej., para valorar el grado de afectación en una enterocolitis necrosante). En el caso de los lactantes podría incluso servir como método alternativo para el diagnóstico de muerte cerebral. También serviría para valorar el grado de inflamación, fibrosis o cicatrización (p. ej., para distinguir lesiones fibróticas o una inflamación en un paciente con enfermedad de Crohn).
Una indicación de esta técnica aprobada por la Food and Drug Administration en niños es su empleo en la urosonografía de eliminación, para el diagnóstico del reflujo vesicoureteral, como una alternativa a la cistourografía convencional. La técnica consiste en la administración de las microburbujas a través de una sonda urinaria y evaluar la existencia de reflujo con la ecografía. Su mayor sensibilidad y la posibilidad de evaluar de forma detallada la uretra han posibilitado que reemplace a la cistografía convencional.
Elastografía
La elastografía es una técnica de imagen funcional que valora la rigidez de los tejidos determinando la velocidad de conducción de las ondas de ultrasonido. Cuanto más rígido es un tejido, más rápido viajan las ondas.
Hay dos tipos de elastografías: de onda cortante (hear-wave) y de tensión o de deformación (strain). La primera se usa para cuantificar el grado absoluto de rigidez de un tejido, y se basa en aplicar ondas de alta intensidad midiendo cómo se propagan lateralmente con la deformación de los tejidos. La segunda es una técnica semicuantitativa de la compresión manual o el movimiento interno fisiológico (p. ej., el latido cardiaco) que resultan en el desplazamiento axial reflejo de la elasticidad del tejido.
Una de las principales ventajas de la elastografía es que ha disminuido la necesidad de realizar biopsias hepáticas. Podría usarse en el diagnóstico de otras patologías, como la detección de un segmento fibrótico en el intestino o de cicatrices renales.
Ecografía en 3 o en 4 dimensiones
Tradicionalmente, el inconveniente de las ecografías en 3 dimensiones (3D) era la duración del procesamiento de las imágenes. Los nuevos avances tecnológicos permiten realizar reconstrucciones de imágenes muy rápidas a partir de los cortes en 2 dimensiones. Son útiles para determinar el volumen de un órgano o de una masa. La ecografía en 4D permite añadir la dimensión tiempo a la ecografía en 3D.
Ecografía Doppler ultrarrápida
Consiste en una técnica Doppler avanzada con una resolución muy alta, muy superior a la convencional. Permite detectar con una sensibilidad 50 veces superior los cambios de flujo en el cerebro, que se correlacionan bien con la actividad neuronal valorada en el electroencefalograma. Esta resolución temporal es muy superior a la de la RM funcional.
Ecografía de alta frecuencia
La ecografía de alta frecuencia proporciona un gran aumento en la resolución, hasta llegar a los 30 µm, superior a la TC o la RM. Se aplica principalmente en neonatología y dermatología, o para el estudio neuromuscular, vascular de tiroides, linfáticos, etc.
Lo que aporta este estudio:
La ecografía ya constituía una herramienta clave en el diagnóstico de todo tipo de lesiones en el paciente pediátrico. Los avances tecnológicos permiten aumentar mucho sus posibilidades no sólo para el diagnóstico, sino también para el seguimiento de las lesiones y, por tanto, como guía en el tratamiento. Ya no sólo proporcionan imágenes, sino que además aportan una información funcional. La ecografía combina facilidad de desplazamiento, bajo precio, ausencia de radiación y sedación innecesaria junto con seguridad, por lo que adquiere una gran relevancia como herramienta en la clínica. Quizás, en un sistema como el español, haya que dilucidar la accesibilidad de esta técnica y la dependencia del radiólogo para su realización. Mientras que otras técnicas de imagen pueden realizarlas los técnicos ante de ser evaluadas por los radiólogos, de momento no ocurre lo mismo con la ecografía. Probablemente, en el marco de la profesión se requiera una reflexión sobre el mejor modo de hacer accesible a la población infantil una herramienta de diagnóstico con tantas virtudes y tan pocos inconvenientes como la ecografía.
J.M. Moreno-Villares
Clínica Universidad de Navarra. Madrid
Estado general de salud y satisfacción vital en niños con enfermedades crónicas
General health and life satisfaction in children with chronic illness
Blackwell CK, Elliott AJ, Ganiban J, Herbstman J, Hunt K, Forrest CB, et al. General health and life satisfaction in children with chronic illness. Pediatrics. 2019; 143(6): e20182988.
Resumen
Los autores de este artículo se plantearon investigar cómo repercuten las enfermedades crónicas en el estado general de salud y en la satisfacción vital de los niños. Gracias a los avances de la medicina y a los cuidados específicos, algunas enfermedades que hace un tiempo eran mortales son actualmente tratables. Se incrementa cada vez más la expectativa de vida, por lo que también se eleva el número de años que se vive con esas enfermedades. La prevalencia de enfermedades crónicas en Estados Unidos y en otros países desarrollados continúa en aumento. Se estima que 1 de cada 5 niños tiene una enfermedad crónica. Los pacientes con estas enfermedades pueden sufrir tanto limitaciones físicas y retrasos del desarrollo psicomotor como dificultades escolares y sociales. De todas formas, aún no se ha determinado el grado de satisfacción vital que tienen estos niños a pesar de su enfermedad.
La satisfacción vital es un componente de la salud y parte de la construcción multidimensional del bienestar, y se define como la evaluación individual de la propia vida como buena y satisfactoria.
Es llamativa la ausencia de estudios sobre satisfacción vital en el contexto de enfermedades crónicas, especialmente en la edad pediátrica.
Los investigadores de este trabajo plantearon dos hipótesis: a) la enfermedad crónica está negativamente asociada con la salud general, y b) la enfermedad crónica no se asocia con la satisfacción vital.
En total, 1.113 cuidadores, pertenecientes a 3 estudios simultáneos de cohortes, completaron el Patient-Reported Outcomes Measurement Information System (PROMIS) para padres con el fin de evaluar el estado general de salud física, mental y social, así como el PROMIS en relación con la satisfacción vital. Participaron 1.253 niños de 5-9 años de edad, entre marzo de 2017 y diciembre de 2017. Un 20% de los niños tenía, al menos, una enfermedad crónica, y un 8,5% más de dos. Los investigadores armonizaron los factores demográficos y los factores ambientales y familiares estresantes (familias monoparentales, salud mental materna e ingresos familiares) con medidas comunes en todas las cohortes. Para examinar las asociaciones entre enfermedades crónicas y el estado de salud de los niños y la satisfacción vital, se ajustaron modelos de regresión lineal.
En cuanto a los resultados del estudio, como era de esperar, los niños con enfermedades crónicas tenían un peor estado general que aquellos sin enfermedades (β ajustada= –1,20; intervalo de confianza [IC] del 95%: –2,49 a 0,09). Sorprendentemente y por contraste, los niños con enfermedades crónicas tenían similares niveles de satisfacción vital (β ajustada= –0,19; IC del 95%: –1,25 a 0,87). En cambio, los niños con diagnóstico psicológico de estrés tenían la asociación negativa más fuerte, tanto para el estado de salud como para la satisfacción vital.
Comentario
La plataforma PROMIS se creó en 2004 por el National Institutes of Health de Estados Unidos para poder evaluar el estado de salud y el resultado de las intervenciones que se realizan al respecto. Es un sistema de información que mide los resultados referidos por los propios pacientes. La valoración del estado de salud, especialmente el de la satisfacción vital, es algo muy personal. Para los investigadores es un reto saber qué es lo que sucede en el interior de los pacientes, cómo valoran su estado de salud y su calidad de vida, y hasta qué grado son consistentes sus respuestas. Esta preocupación ha llevado a la creación de múltiples herramientas de medida del estado de salud y de las intervenciones sociosanitarias. Así surge PROMIS como resultado del trabajo colaborativo de grupos de investigación y centros de reconocido prestigio estadounidenses. Esta plataforma permite generar medidas de resultados referidos por los pacientes de forma estandarizada. Además, se ha hecho el esfuerzo de adaptarlas idiomáticamente para que su uso pueda tener un alcance internacional en la investigación y la práctica clínica. Durante el periodo 2006-2007, el PROMIS recopiló datos de muestras amplísimas, tanto de pacientes como de población general americana, asegurando que existiera una representación adecuada según la edad, el sexo, el nivel educativo, el estatus socioeconómico y el grupo étnico. Con toda esta información se crearon «bancos de ítems», que son herramientas muy útiles para medir aspectos relacionados con la calidad de vida. La plataforma PROMIS cuenta además con el apoyo de modelos matemáticos, que permiten una mayor precisión a la hora de valorar las respuestas de los pacientes. Con estos análisis matemáticos se puede medir el funcionamiento mental humano a la hora de responder a un cuestionario sobre su salud, estableciendo una relación entre el nivel de habilidad del examinado y la probabilidad de que responda correctamente a las preguntas. Los resultados obtenidos son más fiables y se simplifica el estudio, ya que de esta manera los investigadores no necesitan un tamaño muestral tan grande, al contar con herramientas ya validadas y de aplicación universal.
Los autores de este artículo concluyen que, aunque los padres de los niños con enfermedades crónicas valoran con peores resultados la salud de sus hijos, la satisfacción vital de estos niños resulta comparable con la de sus compañeros sanos.
Los niños con enfermedades crónicas aprenden a afrontar y superar situaciones adversas (dolor, sufrimiento, limitaciones funcionales...) y nos dan una lección a los adultos cuando les vemos sonreír y jugar a pesar de sus dificultades.
Lo que aporta este artículo:
Este trabajo de investigación tiene el interés de haber sido realizado a través de la plataforma PROMIS, considerada una herramienta útil y fiable para estudios en relación con el estado de salud y la satisfacción vital, también en la edad pediátrica.
Los progresos de la medicina están facilitando que sobrevivan cada vez más niños con enfermedades graves y crónicas. Con este estudio los autores aportan la evidencia de que las enfermedades crónicas no imposibilitan a los niños tener una vida feliz y satisfactoria.
C. Esteve Cornejo
Pediatra. Clínica Universidad de Navarra. Madrid
Los cereales en la alimentación del lactante y el niño pequeño
Día Mundial de las Enfermedades Raras

Aparecen dos juegos ¿Quién es Quién? gigantes para concienciar sobre las Enfermedades Raras
- En el Día Mundial de estas patologías minoritarias, la Estación de Sants de Barcelona y el Intercambiador de Plaza Castilla de Madrid han amanecido con estas originales instalaciones
- Se trata de una acción de Sanofi Genzyme para visibilizar a las personas que viven con estas enfermedades y reivindicar la importancia de la investigación
Pólipos fibroepiteliales como causa de hidronefrosis
Síndrome de encefalopatía posterior reversible: a propósito de dos casos
El síndrome de encefalopatía posterior reversible es una entidad clinicorradiológica cada vez más conocida en la práctica diaria pediátrica, pero puede presentarse con una sintomatología neurológica muy diversa (cefalea, alteración del nivel de conciencia, pérdida de visión, crisis convulsivas, etc.), generalmente asociada a un cuadro de hipertensión arterial o insuficiencia renal. Para su diagnóstico se utilizan actualmente pruebas de imagen, como la resonancia magnética, con una elevada sensibilidad, la tomografía computarizada y el electroencefalograma. Su manejo radica en el tratamiento de las crisis y el control de la presión arterial, además de la eliminación de los factores predisponentes, y suele tener un buen pronóstico, con una resolución completa del cuadro en días o semanas.
Programa de transición de nefrología pediátrica a la medicina del adulto: «Conduce tu salud»
Lumbalgia en un caso de púrpura de Schönlein-Henoch: de hallazgo incidental a complicación infrecuente
La Real Academia Nacional de Medicina de Madrid acoge la ceremonia de entrega de los 1º Premios AELMHU

Los premiados
La ceremonia de los 1º Premios AELMHU se celebró en la Real Academia Nacional de Medicina de Madrid, con la asistencia de Belén Crespo, directora de la Agencia Española de Medicamentos y Productos Sanitarios, y Mª José Calvo, subdirectora general de Farmacia y Productos Sanitarios de Comunidad de Madrid, quienes, junto al presidente de AELMHU, Josep Mª Espinalt, entregaron los galardones cuyas dotaciones económicas de 3.000 euros han sido donadas íntegramente a proyectos vinculados a las enfermedades raras y/o los medicamentos huérfanos.
La Fundación ALPE Acondroplasia en la categoría de Mejor labor en Difusión; la Dra. Roser Torra, responsable de Enfermedades Renales Hereditarias (ERH) en la Fundació Puigvert de Barcelona, en el apartado de Mejor Trayectoria Clínica; y el Dr. David Araújo, del Hospital Universitario de Santiago (CHUS) en la categoría de Mejor Trayectoria Investigadora, recibieron también como finalistas una placa en el transcurso del acto.
Un total de 33 candidaturas han concurrido a la primera edición de los Premios AELMHU, galardones creados por la Asociación Española de Laboratorios de Medicamentos Huérfanos y Ultrahuérfanos para reconocer los mejores trabajos y trayectorias profesionales en investigación y difusión de las enfermedades raras y los medicamentos huérfanos.
«Para poder contribuir a la búsqueda de soluciones en el ámbito de las enfermedades raras, creemos necesario emprender iniciativas, como estos Premios, que reconozcan la labor de profesionales, asociaciones y empresas por mejorar la vida de quienes padecen una enfermedad poco frecuente y que difundan sus especiales circunstancias, sus retos y el valor terapéutico y social de los medicamentos huérfanos», aseguró el presidente de AELMHU.
AELMHU es una entidad sin ánimo de lucro que agrupa a empresas farmacéuticas y biotecnológicas con un decidido compromiso por descubrir, investigar, desarrollar y comercializar terapias innovadoras capaces de mejorar la situación de los pacientes que padecen enfermedades raras. «Hemos creado estos galardones con una clara proyección de futuro porque creemos que la constancia y la perseverancia de todos los implicados en la investigación y difusión de las enfermedades raras y sus tratamientos debe ser premiada e incentivada», señalo Josep Mª Espinalt.
Jurado
El jurado de los Premios AELMHU, presidido por Dr. Josep Torrent-Farnell, estuvo formado por profesionales clínicos y científicos, expertos en enfermedades raras, representantes de instituciones políticas y periodistas entre los que se encuentra la Dra. Reyes Abad, jefa del Servicio de Farmacia del Hospital Universitario Miguel Servet de Zaragoza; Paloma Casado, subdirectora general de Salud Pública, Calidad e Innovación del Ministerio de Sanidad; Dr. César Hernández, jefe del Departamento de Medicamentos de Uso Humano de la AEMPS; Dr. Pablo Lapunzina, Director científico de CIBERER; Dr. Francesc Palau, director del Servicio de Medicina Genética y Molecular del Instituto Pediátrico de Enfermedades Raras (IPER); Elisabet Pedrosa, periodista de Catalunya Ràdio; Manuel Pérez, presidente del Colegio de Farmacéuticos de Sevilla; Dr. Manuel Posada, director del Instituto de Investigación en Enfermedades Raras (IIER), y Margarita Iniesta, directora ejecutiva de AELMHU, miembro del Jurado sin voz ni voto.
Agenesia del cuerpo calloso como forma de presentación de un síndrome de deleción y duplicación invertida del brazo corto del cromosoma 8
Azul de metileno en el síndrome de escape capilar refractario
Sr. Director:
El síndrome de escape capilar se define como una situación de disfunción endotelial en la que se produce un estado de vasoplejía generalizada y en el que intervienen, entre otros mediadores, el óxido nítrico (NO). Este fallo endotelial se ha descrito en múltiples patologías en el contexto de un síndrome de respuesta inflamatoria sistémica (SIRS) (anafilaxia, sepsis, intoxicación farmacológica, etc.) y hasta en un 10% de los procesos postoperatorios de cirugía cardiovascular1.
En la literatura se encuentran descritos casos de síndrome de escape capilar refractario a drogas y volumen con respuesta favorable a la administración de azul de metileno. La mayor serie en población pediátrica consiste en 5 recién nacidos con shock séptico2.
Presentamos el caso de un recién nacido pretérmino, de 36 semanas de edad gestacional, con una cardiopatía compleja (ventrículo derecho de doble salida con comunicación interventricular y estenosis pulmonar), intervenido a los 26 días de vida mediante una cirugía paliativa consistente en la ampliación del infundíbulo y del tronco de la arteria pulmonar. En el postoperatorio inmediato presentó una situación de hiperaflujo pulmonar y bajo gasto, agravado por un escape capilar grave multifactorial. El paciente presentaba un edema generalizado, presiones de llenado bajas (presión venosa central <8) a pesar de un aporte continuo de volumen (>50 mL/kg), soporte vasoconstrictor en dosis altas (noradrenalina hasta 0,64 µg/kg/min), hidrocortisona a 25 mg/m2, resistencias vasculares sistémicas bajas (<1.000) medidas con monitor MostCare®, y anuria mantenida. Como factores predisponentes para desarrollar un síndrome de escape capilar cabe citar la utilización de prostaglandinas prequirúrgicas en dosis altas, el bypass cardiopulmonar prolongado (157 min), la prematuridad del recién nacido y haber recibido un tratamiento betabloqueador previo3.
Ante la refractariedad de la situación, se decidió administrar azul de metileno en perfusión en 1 hora, a 1 mg/kg, con lo que mejoró de manera franca la presión arterial del paciente; las presiones de llenado se mantuvieron estables sin necesidad de aporte continuo de volumen, se inició la diuresis y pudo disminuirse el soporte vasoconstrictor en el plazo de 6 horas a 0,4 µg/kg/min. No se identificaron efectos secundarios atribuibles al tratamiento con azul de metileno, salvo la coloración azul de la piel, las lágrimas y la orina.
El azul de metileno es un compuesto químico cuyo uso como medicamento se inició en 1800, inicialmente como antimalárico. Ahora es bien conocida su utilidad como marcador en la cirugía oncológica, como agente reductor en la metahemoglobinemia y, más recientemente, como competidor del NO en el músculo liso vascular4.
En la síntesis del NO en el endotelio y el músculo liso vascular intervienen las enzimas óxido nítrico sintetasa endotelial (eNOs) y la óxido nítrico sintetasa inducible (iNOs). Éstas producen NO, que induce a su vez la guanilciclasa sintetasa (sGC), que sintetiza GMPc, mediador celular de muchos procesos, entre ellos la relajación del músculo liso vascular. En el SIRS se sabe que la liberación de citocinas inflamatorias actúa induciendo la iNOs, lo que conduce a una síntesis excesiva de NO que supera la capacidad del endotelio de mantener un adecuado tono vascular.
El azul de metileno, por su papel como competidor del NO, consigue inhibir la síntesis de sGC y disminuir los depósitos de NO; así pues, se comprende su papel en el tratamiento de la vasoplejía. Su uso está recomendado en la misma dosis que en la metahemoglobinemia (1-2 mg/kg), que según se describe en otras series, puede repetirse o incluso administrarse en perfusión continua (no fue necesario en este paciente).
Se trata de un fármaco con un perfil bastante seguro, contraindicado en situaciones de insuficiencia renal grave, déficit de G6PDH o hipersensibilidad a las dapsonas. Como efectos secundarios más graves, aunque raros, se han descrito los siguientes: arritmias, vasoconstricción coronaria, alteración del intercambio gaseoso por vasoconstricción pulmonar y encefalopatía aguda5.
El azul de metileno puede ser útil como tratamiento de rescate del síndrome de escape capilar cuando exista una refractariedad al tratamiento convencional. Dado que no hay estudios que avalen su uso como tratamiento de primera línea con esta indicación, y hasta disponer de una evidencia de mayor calidad, debe utilizarse con un adecuado conocimiento de sus indicaciones, contraindicaciones y potenciales efectos secundarios, aunque algunos autores, como Evora et al.6, defienden su uso precoz.
Bibliografía
1. Shanmugam G. Vasoplegic syndrome: the role of methylene blue. Eur J Cardiothorac Surg. 2005; 28(5): 705-710.
2. Driscoll W, Thurin S, Carrion V, Steinhorn RH. Effect of methylene blue on refractory neonatal hypotension. J Pediatr. 1996; 129(6): 904-908.
3. Omar S, Zedan A, Nugent K. Cardiac vasoplegia syndrome: pathophysiology, risk factors and treatment. Am J Med Sci. 2015; 349(1): 80-88.
4. Jang DH, Nelson LS, Hoffman RS. Methylene blue for distributive shock: a potential new use of an old antidote. J Med Toxicol. 2013; 9(3): 242-249.
5. Lo JC, Darracq MA, Clark RF. A review of methylene blue treatment for cardiovascular collapse. J Emerg Med. 2014; 46(5): 670-679.
6. Evora PR, Alves Junior L, Ferreira CA, et al. Twenty years of vasoplegic syndrome treatment in heart surgery. Methylene blue revised. Rev Bras Cir Cardiovasc. 2015; 30(1): 84-92.
Daño renal agudo debido a un síndrome de lisis tumoral espontáneo como primera manifestación del linfoma de Burkitt
El síndrome de lisis tumoral espontáneo (SLTE) es una causa excepcional de daño renal agudo (DRA) en la infancia. Describimos un caso de DRA secundario a SLTE como primera manifestación de un linfoma de Burkitt. Se trata de un niño de 5 años de edad, con anorexia de 1 mes de evolución, niveles sanguíneos elevados (mg/dL) de urea/creatinina (337/7,21), ácido úrico (30,4) y fósforo (7), y unas cifras de potasio de 6,6 mEq/L, que presenta unos riñones grandes e hiperecogénicos en la ecografía abdominal. Experimentó una mejoría progresiva de la función renal tras el inicio de tratamiento con rasburicasa e hiperhidratación. El día +14 empeoró clínicamente; se repitió la ecografía y se detectó una gran masa retroperitoneal, por lo que se realizó una biopsia. Durante el procedimiento, tras administrar dexametasona (protocolo de intubación), presentó taquicardia ventricular secundaria a hiperpotasemia (9 mEq/L), que revirtió sin cardioversión. Precisó hemofiltración durante 48 horas. Tras el diagnóstico anatomopatológico de linfoma de Burkitt, se inició un tratamiento específico, y actualmente el paciente está en remisión. Ante un caso de DRA con hiperuricemia, hiperfosforemia e hiperpotasemia, debemos sospechar un SLTE, descartar un proceso tumoral oculto y evitar la administración de esteroides, ya que puede resultar catastrófica.
Síndrome nefrítico agudo asociado a infección por «Mycoplasma pneumoniae»
Evitar el legado de la enfermedad renal. Enfoque sobre la niñez
El Día Mundial del Riñón 2016 se centra en las enfermedades renales durante la infancia y la enfermedad renal del adulto que puede iniciarse en una edad temprana. La enfermedad renal crónica (ERC) en la infancia difiere de la de los adultos en que predominan las causas derivadas de anomalías congénitas y trastornos hereditarios; las glomerulopatías y la enfermedad renal asociada a diabetes mellitus son poco frecuentes. Además, muchos niños con una lesión renal aguda pueden desarrollar eventualmente secuelas que lleven a la hipertensión y la ERC durante la adolescencia o la vida adulta. Los niños nacidos prematuros o pequeños para su edad gestacional tienen un incremento del riesgo de desarrollar ERC durante su vida. Las personas con un alto riesgo al nacer o en la primera infancia deberán ser monitorizadas estrechamente para poder detectar a tiempo los signos tempranos de enfermedad renal, con el fin de proporcionar una prevención o un tratamiento efectivos. Es factible establecer una terapia eficaz en niños con ERC avanzada; existen evidencias que demuestran que los niños evolucionan mejor que los adultos cuando reciben terapia de reemplazo renal, incluyendo la diálisis y el trasplante, aunque sólo una minoría requiere este tratamiento. Debido a las inequidades en el acceso a la atención médica, es necesario hacer un esfuerzo para que los niños con una enfermedad renal, donde sea que vivan, puedan ser tratados de manera eficaz, independientemente de su ubicación geográfica y su situación económica. Nuestra esperanza es que el Día Mundial del Riñón pueda informar al público en general, a los gestores y a los profesionales de la salud sobre las necesidades y las posibilidades que existen en torno a la enfermedad renal en la infancia.
Presentación de un síndrome hiponatrémico-hipertensivo como síndrome de poliuria-polidipsia
El síndrome hiponatrémico-hipertensivo es infrecuente en niños. Se caracteriza por la presencia de hipertensión de origen renovascular y trastornos hidroelectrolíticos, asociado a hiponatremia, que conduce a una depleción de sodio y agua, lo que se define como natriuresis por presión. La mayoría de los casos publicados en los pacientes pediátricos presentan síntomas neurológicos agudos acompañados de vómitos, retraso del crecimiento y pérdida de peso. Sólo D’Angelo et al. describieron 2 casos similares con SHH y síndrome poliuria-polidipsia, pero ambos asociados a un tumor de Wilms. Presentamos el caso de un niño de 2 años de edad con síndrome hiponatrémico-hipertensivo, encontrado durante un estudio de poliuria-polidipsia. Aunque los valores de presión arterial eran extremadamente altos, el paciente no presentaba clínica neurológica. Los niveles de actividad de renina plasmática y aldosterona estaban elevados. La arteriografía reveló la existencia de una estenosis de una arteria renal accesoria del riñón derecho. Tras un tratamiento antihipertensivo intensivo, el paciente pudo someterse a cirugía, alcanzando posteriormente valores normales de presión arterial y desapareciendo la poliuria y la polidipsia. Este caso supone una presentación del síndrome hiponatrémico-hipertensivo no descrita hasta ahora, y confirma que la hipertensión renovascular puede manifestarse sólo como un síndrome de poliuria-polidipsia a pesar de presentar valores extremadamente elevados de presión arterial.
Glomerulonefritis aguda secundaria a una infección por parvovirus B19
El parvovirus B19 es un virus patógeno humano. Aunque su asociación al daño renal es rara en la edad pediátrica, se han descrito algunos casos de glomerulonefritis aguda en el contexto de una infección por parvovirus B19. Describimos el caso de una niña de 12 años de edad, previamente sana, que desarrolló edemas y proteinuria. Los estudios serológicos determinaron anticuerpos IgM antiparvovirus B19. El genoma del parvovirus B19 fue detectado mediante reacción en cadena de la polimerasa. La paciente se recuperó espontáneamente, con desaparición de la proteinuria y resolución de la hipocomplementemia. La infección por parvovirus B19 se ha asociado al daño renal y es uno de los agentes etiológicos de glomerulonefritis aguda.
En Noviembre de 1964 «Acta Pediátrica Española» publicaba...
FIGURA DE LA PEDIATRÍA
ARTÍCULOS ORIGINALES
El síndrome de la trisomía autosómica 17-18, por los doctores José Luis Bezanilla y María Carmen Elorza
Oligofrenia, convulsiones y argininuria, por el doctor Ángel Peralta Serrano
Aminoaciduria en las enfermedades renales de la infancia, por los doctores M. Cruz Hernández, C. Gilabert y J.A. Molina Font
Otitis media del lactante, por el doctor Bernardo Pérez Moreno
Rabdomiolisis aguda: revisión y evaluación del daño renal
Introducción: Analizamos a los pacientes atendidos en nuestro hospital con rabdomiolisis, valorando su función renal y las características relacionadas.
Material y métodos: Estudio retrospectivo de 2,5 años, en el que se incluyen pacientes menores de 16 años con cifras de creatinfosfocinasa (CPK) >1.000 UI/L. Se excluyeron los menores de 1 mes y los que presentaban una elevación de CPK de origen cardiaco. Definimos daño renal agudo según los criterios RIFLE adaptados a pediatría. Se recogieron diferentes variables clínicas y bioquímicas.
Resultados: Se analizaron 55 pacientes (mediana de edad de 8 años) con CPK inicial de 1.826 UI/L (rango: 1.213-4.414). Las causas más frecuentes fueron las miositis virales, la cirugía muscular y las convulsiones. El 15,9% presentó daño renal agudo, que no se relacionó con la causa de rabdomiolisis y se asoció a cifras elevadas de CPK. Ninguno precisó depuración extrarrenal.
Conclusiones: El daño renal agudo asociado a rabdomiolisis suele ser leve y más frecuente en los niños con valores más elevados de CPK.
II Reunión Nacional de Nefrourología Pediátrica. Puesta al día en el diagnóstico prenatal de anomalías estructurales del riñón y las vías urinarias
Objetivo: Precisar las indicaciones quirúrgicas y la realización de pruebas diagnósticas en pacientes con anomalías estructurales del riñón y las vías urinarias de diagnóstico prenatal.
Material y métodos: Se ha revisado la bibliografía más reciente y se han comparado los resultados con la encuesta enviada a los 108 inscritos en la II Reunión Nacional de Nefrourología Pediátrica (nefrólogos y urólogos pediátricos, principalmente) sobre sus pautas de actuación. Se obtuvieron 30 respuestas.
Resultados: Casi el 90% de las hidronefrosis de diagnóstico intraútero son transitorias. Los pacientes con un diámetro anteroposterior de la pelvis renal en ecografía <15 mm, realizada no antes del tercer día de vida, no deben ser objeto de pruebas invasivas. No se recomienda realizar una cistografía a todos los niños con dilatación de manera sistemática. En el renograma diurético, la pérdida de función renal en sucesivos renogramas es el principal indicador de intervención. Se recomienda realizar profilaxis antibiótica en pacientes de riesgo.
Conclusión: Las respuestas a la encuesta coinciden mayoritariamente con lo recomendado en la bibliografía. El plan inicial con estos pacientes debe ser mínimamente invasivo. Los estudios deben realizarse en el momento del nacimiento en caso de sospecha de obstrucción bilateral o de vía común. En caso de dilataciones unilaterales, la evaluación debe efectuarse a partir del tercer día de vida. Recomendamos realizar profilaxis antibiótica, al menos hasta finalizar los estudios, en las hidronefrosis graves.
Golpe de calor en un niño en tratamiento con metilfenidato y topiramato
Entre las entidades patológicas relacionadas con el calor, el denominado «golpe de calor», tanto clásico como relacionado con el ejercicio, es el más grave, y representa una amenaza para la vida del paciente. Esta entidad se define como una elevación de la temperatura corporal central mayor de 40 ºC acompañada de signos de disfunción neurológica (confusión, ataxia, disminución del nivel de conciencia, convulsiones o coma). Las complicaciones pueden ser graves, e incluyen las siguientes: fallo renal agudo, el fallo hepático, rabdomiolisis, colapso cardiovascular, alteraciones hidroelectrolíticas, trombocitopenia, coagulación intravascular diseminada y fallo multiorgánico. El golpe de calor se produce cuando los mecanismos de adaptación de la pérdida de calor se ven abrumados, lo que provoca un desequilibrio entre la producción y la pérdida de calor. Además, existen factores intrínsecos (edad, sexo, enfermedad crónica, medicación, deshidratación, obesidad...) y extrínsecos (temperatura y humedad ambiental, ejercicio físico...) que pueden contribuir al desarrollo de un golpe de calor. El tratamiento se basa en el enfriamiento inmediato, el soporte de la función sistémica y la prevención de las complicaciones. El pronóstico depende de la severidad de la afectación del sistema nervioso central, así como de la intensidad y la duración de la hipertermia.
Presentamos el caso de un varón de 13 años en tratamiento con topiramato y metilfenidato, que sufrió un golpe de calor mientras realizaba ejercicio en un ambiente caluroso, y describimos la sintomatología que compone esta entidad.
Afectación renal en las cardiopatías congénitas
Las cardiopatías congénitas (CC) son las anomalías congénitas estructurales más frecuentes, y pueden asociar problemas renales congénitos de forma no desdeñable. Aunque la afectación cardiaca secundaria a problemas renales está bien determinada, los efectos que producen las CC en sus distintas variantes sintomáticas sobre los riñones son bastante desconocidos. La mejora de los medios técnicos y humanos desde los comienzos en esta área hasta nuestros días ha permitido el aumento de la supervivencia y la calidad de vida a medio-largo plazo de estos pacientes. Esto ha diversificado la morbilidad asociada a su evolución y tratamiento, y es menor el daño derivado de la evolución natural de las CC. Existe una mayor concienciación sobre la profilaxis y el tratamiento precoz de los problemas derivados, fundamentalmente, de la cirugía con circulación extracorpórea, las cirugías paliativas y el trasplante cardiaco. Asimismo, se tiene un mayor conocimiento y se toman más precauciones en la administración de los múltiples fármacos con efectos adversos renales que pueden usarse en los pacientes con CC.
Obstrucción urinaria por cálculo uretral
La litiasis renal es una patología típica del adulto, y menos frecuente en la edad pediátrica, por lo que no siempre se piensa en ella. Presentamos el caso de un niño de 3 años de edad con un cálculo uretral, previamente diagnosticado de infección de orina, que acudió al servicio de urgencias con síntomas de obstrucción urinaria, provocados por la expulsión en ese momento del cálculo.
Hidrometrocolpos, polidactilia postaxial y anomalías cardiacas: el síndrome de McKusick-Kaufman como reto diagnóstico prenatal y posnatal
El síndrome de McKusick-Kaufman se caracteriza por la presencia de hidrometrocolpos, polidactilia postaxial y anomalías cardiacas en las mujeres, y malformaciones genitales en los varones. Mostramos el caso de una recién nacida que presentaba una gran masa quística intraabdominal e hidronefrosis bilateral en la ecografía practicada en la semana 32 de gestación. Tras el nacimiento, se confirmó dicha masa quística, así como la existencia de hidrometrocolpos, polidactilia postaxial y comunicación interauricular, por lo que fue diagnosticada de síndrome de McKusick-Kaufman.
Agenesia sacra tipo I: diagnóstico a través de la exploración
La agenesia sacra es una malformación poco frecuente que forma parte del síndrome de regresión caudal. Presentamos el caso de una recién nacida que muestra en la exploración física una desviación del surco interglúteo, una fosita lumbar y máculas hipocrómicas a la altura del sacro, que hacen sospechar una anomalía congénita lumbosacra. Se realizan estudios de imagen (radiografía, ecografía y resonancia magnética) que confirman el diagnóstico de agenesia sacra tipo I. La paciente presenta, a su vez, una displasia congénita de cadera izquierda que precisó una férula de Pavlik para su corrección. Su evolución fue favorable, manteniéndose asintomática hasta el momento actual.
Papel de los cereales en la alimentación infantil
En el primer año de vida, la lactancia materna es el referente durante al menos los 6 primeros meses, pero hay una cierta dispersión de tendencias cuando se llega a la edad de introducir la alimentación complementaria. Tradicionalmente, los cereales han sido y son los primeros alimentos que se aconsejan como inicio de la alimentación complementaria. En los últimos tiempos se ha observado un descenso que posiblemente esté relacionado con algunos tópicos negativos, por lo que nos ha parecido oportuno actualizar su papel.
Las características fisiológicas de los primeros años de vida son circunstancias que tienen gran importancia para conseguir una buena adaptación en la progresión de la alimentación atendiendo a las capacidades que el nuevo ser va adquiriendo en estos primeros años, preferentemente en sus funciones digestivas, renales y neuromusculares.
Los cereales son una excelente fuente nutricional, pero no todos tienen las mismas propiedades, por lo que se repasa su composición para valorar los beneficios que representan en la salud del niño, tanto como aporte de la energía que va necesitando para cubrir sus necesidades, como para evitar deficiencias proteicas y de vitaminas y oligoelementos. No sólo es importante la cantidad, sino también la calidad y el contexto de una alimentación variada y equilibrada que ayude a compensar el resto de los aportes.
Ya que los cereales son fundamentales en la alimentación de nuestros hijos, es bueno recordar algunas recomendaciones actualizadas sobre su uso en los diferentes periodos de la vida, ya sea en forma de papillas en los primeros meses o de cereales más complejos en etapas más avanzadas, así como resaltar la vigencia que conservan en la alimentación de los primeros años si se utilizan correctamente.
Nefroma mesoblástico congénito con hiperecogenicidad medular que simula nefrocalcinosis
El nefroma mesoblástico congénito (NMC) es un tumor raro, siendo el más frecuente a nivel renal en los pacientes menores de 2 meses. Su origen histológico es la estroma renal inmadura, y se distinguen los subtipos clásico, mixto y celular. El tratamiento de elección es quirúrgico y su pronóstico es excelente. Se han descrito casos de NMC asociado a nefrocalcinosis en relación con la hipercalcemia paraneoplásica. Exponemos el caso de un recién nacido que presenta en la ecografía imágenes de hiperecogenicidad medular renal bilateral, similar a una nefrocalcinosis, en el contexto clínico de un NMC.
Distrofia torácica asfixiante, o síndrome de Jeune, asociada a una malformación espinal cervical
La distrofia torácica asfixiante, o síndrome de Jeune, es una displasia ósea de herencia autosómica recesiva, con expresión fenotípica variable. El diagnóstico es fundamentalmente clínico y radiológico. Se caracteriza por la presencia de un tórax estrecho y acampanado, polidactilia, costillas horizontalizadas y huesos iliacos cortos, con una alteración típica del techo acetabular en tridente, y suele asociar otras complicaciones: nefrocalcinosis, hepatopatía colestásica, anomalías pancreáticas y retinianas. La distrofia torácica produce hipoplasia pulmonar secundaria e insuficiencia respiratoria restrictiva que puede ser mortal en etapas precoces de la vida. Aunque la existencia de malformaciones espinales no es un hallazgo frecuente, en este caso, el paciente presentaba una malformación de C1 que producía compresión medular. Se realizó una descompresión quirúrgica y, posteriormente, la evolución del paciente ha sido muy favorable. Concluimos que debe realizarse siempre el cribado de malformaciones espinales en pacientes afectados de este síndrome, ya que su tratamiento puede contribuir significativamente a mejorar su pronóstico y calidad de vida.
Convulsión hiponatrémica con parada respiratoria de etiología poco común
Las crisis convulsivas pueden ser la primera manifestación de una patología insospechada. Junto con el tratamiento anticonvulsivo, es necesario un protocolo de actuación dirigido a descartar y tratar las posibles causas reversibles. Las convulsiones por hiponatremia son una manifestación de máxima gravedad con claro riesgo vital, por lo que es necesaria una rápida actuación que eleve la natremia y la osmolaridad a cifras de seguridad. Una causa infrecuente de hiponatremia en los lactantes es la pérdida renal de sodio, secundaria a una infección del tracto urinario. Su origen parece deberse a una resistencia a la acción de la aldosterona en los túbulos renales.
Presentamos el caso clínico de un lactante de 49 días de vida, que presentó una crisis convulsiva y una parada respiratoria en el contexto de una hiponatremia grave (Na+: 110 mEq/L). El urocultivo confirmó una pielonefritis por Enterobacter aerogenes, y la ecografía una pielocaliectasia bilateral con cistografía normal, sin evidencia de reflujo vesiculoureteral.
Seudohipoparatiroidismo neonatal transitorio: una causa rara de hipocalcemia neonatal tardía
El seudohipoparatiroidismo neonatal transitorio es un cuadro escasamente descrito, que cursa con hipocalcemia neonatal tardía, hiperfosfatemia y niveles elevados de hormona paratiroidea (PTH), lo que refleja resistencia periférica a su acción. Es una causa infrecuente de hipocalcemia neonatal tardía, y el defecto bioquímico parece residir en una inmadurez funcional de los receptores renales de la PTH. Para su corrección, se precisan aportes elevados de calcio y análogos de vitamina D. Su carácter autolimitado lo diferencia de otros seudohipoparatiroidismos persistentes. Exponemos el caso de una recién nacida pretérmino, con crecimiento intrauterino retardado, que presentó esta patología. Analizaremos los hallazgos clínicos y bioquímicos, así como el diagnóstico diferencial y el manejo de este raro trastorno.
Guía clínica para el diagnóstico, tratamiento y seguimiento de la enfermedad de Gaucher en la infancia
Introducción
La enfermedad de Gaucher, con una frecuencia media de uno por cada 40.000 recién nacidos como mínimo, debuta en más de la mitad de los pacientes antes de los 18 años. Cuando se inicia en la infancia suele tener una evolución clínica más rápida y grave que en la edad adulta, existe una relación directa entre el tratamiento precoz y la adecuada respuesta terapéutica, y es muy probable que la aparición de algunas importantes manifestaciones clínicas de la enfermedad necesiten ser prevenidas durante la infancia. Por ello, resulta fundamental aplicar lo antes posible las medidas diagnósticas adecuadas ante todo enfermo con sintomatología compatible con enfermedad de Gaucher.
El diagnóstico y tratamiento de esta enfermedad pueden presentar dificultades debido a su gran variabilidad clínica. Por ello, las recomendaciones que se recogen a continuación son de carácter general y en todos los casos el pediatra responsable del paciente debe individualizar las medidas diagnósticas, terapéuticas o de seguimiento en función de las necesidades de cada niño a lo largo del tiempo y de acuerdo con las variaciones que se vayan produciendo en el conocimiento de la enfermedad.
Con el fin de recibir la mejor asistencia sanitaria posible, todo paciente afectado debe ser asistido por un equipo multidisciplinario en un centro pediátrico con experiencia en el tratamiento de enfermedades metabólicas en la infancia.
Diagnóstico
Sospecha clínica
Se basa en la presencia aislada o en combinación de los siguientes signos o síntomas:
• Astenia.
• Retraso del crecimiento.
• Retraso de la maduración sexual.
• Palidez, petequias, equimosis, sangrado «espontáneo».
• Esplenomegalia, hepatomegalia.
• Dolor abdominal, distensión abdominal.
• Alteraciones esqueléticas: osteopenia, osteoporosis, osteonecrosis, dolor óseo agudo, fracturas patológicas, lesiones líticas, deformidades esqueléticas.
• Alteraciones cutáneas: hidropesía fetal, recién nacido colodión, ictiosis congénita.
• Alteraciones del sistema nervioso central (SNC): alteración de los movimientos sacádicos, estrabismo, ataxia, trismo, epilepsia mioclónica, deterioro intelectual.
Analítica compatible
Los siguientes hallazgos analíticos apoyan fuertemente el diagnóstico:
• Anemia. Trombocitopenia.
• Fosfatasas ácidas tartratorresistentes (TRAP) elevadas.
• Quitotriosidasa elevada.
• Presencia de células de Gaucher en el aspirado medular.
Confirmación diagnóstica
La certeza diagnóstica requiere en todos los casos (incluso en los hermanos de los afectados):
• Comprobación de una actividad de la β-glucocerebrosidasa disminuida en leucocitos, fibroblastos u otras células nucleadas del paciente.
Comentarios
Se han subrayado los datos más significativos para el diagnóstico, por su frecuencia o importancia.
Evaluación inicial del paciente
Examen clínico
Antecedentes familiares
• Etnia (si procede, por la epidemiología de la enfermedad).
• Genealogía familiar.
• Peso, talla y perímetro craneal de padres y hermanos.
• Antecedentes patológicos (hemopatías, Parkinson, demencia).
Antecedentes personales
• Embarazo, parto, periodo neonatal.
• Desarrollo psicomotor.
• Crecimiento.
Enfermedad actual
• Falta de medro.
• Astenia, polipnea.
• Dolor o distensión abdominal.
• Palidez, hematomas, sangrado mucoso.
• Dolor óseo, fracturas.
• Alteración del comportamiento social.
• Disminución del rendimiento escolar.
• Cualquier síntoma neurológico.
Examen físico
• Peso, talla, perímetro craneal, índice de masa corporal (IMC) (percentiles).
• Estadio de desarrollo puberal (Tanner).
• Palidez, equimosis, petequias.
• Esplenomegalia. Hepatomegalia.
• Examen de la motilidad ocular.
• Examen de audición.
• Examen neurológico.
• Examen cardiovascular.
• Examen del aparato respiratorio.
• Examen del sistema esquelético.
Exámenes complementarios
• Hematología:
– Hemograma. Metabolismo del hierro.
– Pruebas de coagulación.
• Bioquímica:
– Perfil lipídico.
– Calcio, fósforo, fosfatasas alcalinas, proteínas, albúmina.
– Función hepática. Función renal.
– Fosfatasa ácida tartratorresistente, quitotriosidasa.
• Biología molecular:
– Examen de mutaciones del gen codificador de la glucocerebrosidasa.
• Diagnóstico por imagen:
– Examen del esqueleto: radiología simple de fémur, tibia, columna y áreas sintomáticas; radiografía (Rx) de carpo izquierdo; Rx de tórax; resonancia magnética (RM) T1 (médula) y T2 (estado vascular) ponderadas de fémur y tibia.
– Densitometría ósea: absorciometría de rayos X con energía dual (DEXA) o ultrasonidos.
– Volumen de hígado y bazo: cuantificado mediante RM o ecografía.
• Electrooculografía.
– Examen de los movimientos sacádicos (videoculografía).
• Oftalmoscopia.
• Electrofisiología:
– Electroencefalografía (EEG) y potenciales evocados tronculares (PET).
• Examen audiométrico:
– Audiometría.
• RM cerebral y troncular.
– Si existe enfermedad neurológica o en pacientes con sospecha de alteración neurológica y con mutación génica «de riesgo».
• Examen de la función pulmonar.
• Ecocardiografía.
• Desarrollo intelectual:
– Brunet-Lezine (o Bayley II) <3 años.
– McCarty 3-7 años.
– Wechsler >7años.
• Valoración de la calidad de vida.
Comentarios
La evaluación inicial es muy importante ya que permite definir el grado de riesgo del paciente y seguir de forma adecuada su evolución. La práctica de algunos exámenes complementarios que precisan colaboración activa vendrá condicionada por la edad del niño, pero en todos los casos se realizará un examen lo más riguroso y minucioso posible.
El examen de la función pulmonar puede efectuarse mediante ecocardiografía para medir el gradiente transpulmonar por debajo de los 4 años y, por encima de esta edad, puede usarse ecocardiografía, espirometría o pletismografía. La alteración de la función pulmonar no es habitual durante la primera década de la vida.
En el caso de que exista la posibilidad de utilizar diferentes métodos de exploración, se utilizará aquel para el que se posea más experiencia y se procurará que sea siempre el mismo a lo largo del tiempo.
Para la evaluación de la calidad de vida debe usarse el cuestionario expresamente confeccionado y validado para la infancia: «Versión española del cuestionario PedsQL aplicado a niños con enfermedad de Gaucher».
Definición de los objetivos terapéuticos
El objetivo del tratamiento es recuperar al paciente de los síntomas presentes y evitar las posibles manifestaciones futuras. En pediatría, los aspectos preventivos alcanzan su máximo significado y, por ello, es muy importante definir los objetivos terapéuticos de un modo que permita su cuantificación y valoración de forma objetiva a lo largo del tiempo.
• Hemoglobina (Hb):
– Aumentar la Hb a 11 g/dL a los 12-24 meses de tratamiento.
• Plaquetas:
– Aumentar las plaquetas para evitar el sangrado durante el primer año. En la trombocitopenia moderada, incrementar las plaquetas de 1,5 a 2 veces durante el primer año y alcanzar el nivel mínimo normal al segundo año de tratamiento.
– En la trombocitopenia grave, aumentar las plaquetas 1,5 veces el primer año e incrementar lentamente durante los años 2 a 5 (lo ideal es que durante el segundo año se dupliquen), aunque no se normalice la cifra.
– Evitar la esplenectomía.
– Mantener la máxima cifra alcanzada sin sangrado de un modo estable.
• Hepatomegalia:
– Reducir y mantener el volumen, como máximo, entre 1 y 1,5 veces su valor normal.
– Disminuir el volumen un 20-30% en los años 1 y 2 de tratamiento, y un 30-40% en los años 3 a 5.
• Esplenomegalia:
– Reducir y mantener el volumen, como máximo, entre 1 y 1,5 veces su valor normal.
– Reducir el volumen un 20-30% en los años 1 y 2 de tratamiento, y un 30-40% en los años 3 a 5.
• Alteraciones óseas:
– Eliminar el dolor óseo en 1-2 años. Evitar crisis de dolor óseo.
– Evitar osteonecrosis y colapso articular subcondral.
– Alcanzar el pico de masa ósea ideal para su edad.
– Aumentar el espesor cortical y la densidad mineral trabecular en 2 años.
• Crecimiento y maduración:
– Alcanzar la talla normal para su edad a los 3 años de tratamiento.
– Alcanzar un desarrollo puberal normal.
Comentarios
Estos objetivos son «mínimos» y generales para todos los niños. Con independencia de ellos, cada paciente puede precisar más objetivos en función de sus manifestaciones clínicas iniciales.
El hígado supone en varones normales de entre 5 y 12 años el 3,5% del peso corporal y el 2,5% después de esta edad. En mujeres, los valores normales son del 3,2 y el 2,9%, respectivamente. La equivalencia aproximada es de 1 gramo por cada centímetro cúbico de volumen hepático.
El bazo supone, aproximadamente, el 0,2% del peso corporal y la equivalencia aproximada es de 0,45-0,6 g por cada centímetro cúbico de volumen.
No se incluyen objetivos terapéuticos para las manifestaciones neurológicas de la enfermedad porque no existe, por el momento, un tratamiento que permita su manejo ni monitorizar la evolución a largo plazo.
Definición individual del riesgo
Una vez establecidos los objetivos terapéuticos, los pacientes pueden definirse en función del grado de riesgo que tienen para desarrollar una evolución clínica grave y, por tanto, seleccionar el tipo de tratamiento inicial de acuerdo con los datos obtenidos de la evaluación basal.
Enfermedad sin manifestaciones neurológicas
• Riesgo alto:
– Enfermedad que produce síntomas subjetivos (astenia, anorexia, dolor, etc.).
– Retraso de crecimiento.
– Evidencia de afectación esquelética.
– Plaquetas <60.000/mm3 y/o hemorragia «anómala».
– Hemoglobina <2 g/dL, por debajo del valor normal para la edad.
– Afectación de la calidad de vida.
– Hermano afecto de enfermedad de Gaucher grave.
• Riesgo normal:
– Cualquier niño con déficit de b-glucuronidasa y síntomas clínicos presentes.
Enfermedad con manifestaciones neurológicas
Todos los niños con síntomas neurológicos secundarios a la enfermedad de Gaucher son niños de alto riesgo para la evolución clínica.
Comentarios
La clasificación de alto o bajo riesgo se hace a priori y, por lo tanto, puede ocurrir que la evolución del paciente esté en contradicción con ella. En todo momento el pediatra deberá ajustar su conducta a las necesidades individuales de su paciente.
Tratamiento
El tratamiento de la enfermedad de Gaucher viene condicionado por la presencia o no de manifestaciones neurológicas.
Tratamiento del paciente sin patología neurológica
• Pacientes tributarios de tratamiento:
– Todos los pacientes con síntomas deben recibir tratamiento, con independencia de su edad o la gravedad de las manifestaciones clínicas.
• Tipo de tratamiento:
– En el momento actual, por debajo de los 18 años, el tratamiento debe efectuarse en todos los casos con glucocerebrosidasa manosa-terminal recombinante (Cerezyme®) por vía intravenosa (tratamiento enzimático sustitutivo [TES]).
• Dosis:
– Niños de alto riesgo: iniciar 60 U/k cada 2 semanas. A los 3 meses, iniciar la valoración de la respuesta terapéutica.
– Niños de bajo riesgo: iniciar 30-60 U/k cada 2 semanas. A los 3 meses, iniciar la valoración de la respuesta terapéutica.
• En todos los casos:
– Se recomiendan aumentos o reducciones de dosis en fracciones de 30 unidades.
– No reducir la dosis antes de los 12-18 meses de tratamiento. A partir de ese momento, se puede valorar la dosis a utilizar cada 6 meses.
– Dosis mínima a utilizar: 30 U/k cada 15 días.
– No retirar el TES durante la infancia.
Tratamiento del paciente con patología neurológica
Tratamiento de la forma crónica neuronopática
• Pacientes tributarios de tratamiento:
– Forma crónica neuronopática identificada: niños con enfermedad tipo 3 comprobada.
– Niños con riesgo de desarrollar la forma crónica neuronopática: 1. Niños con enfermedad de Gaucher que son hermanos de niños con tipo 3 comprobada; 2. Niños con genotipos de «riesgo»; por ejemplo: L444P/L444P, D409H/D409H, L444P/D409H, etc., y 3. Niños con inicio de la enfermedad antes de los 2 años de edad y síntomas clínicos graves.
• Tipo de tratamiento:
– Hoy en día el único tratamiento farmacológico autorizado en este tipo de pacientes es el uso del TES. Los resultados son poco efectivos en la mayoría de los casos, pero los pacientes con esta forma clínica deben beneficiarse de un intento terapéutico.
– El tratamiento de esta forma de la enfermedad con inhibidores de la síntesis de la glucosilceramida y TES está en fase de ensayo clínico y todavía no se conocen los resultados definitivos.
– Puede considerarse el trasplante de médula o de células de cordón procedentes de donante no emparentado cuando no se obtenga buena evolución con el TES en estos pacientes. Esta opción terapéutica, aunque no está definitivamente descartada, cada vez es menos utilizada porque el balance entre el riesgo y los beneficios obtenidos no parece muy positivo en la mayoría de los casos.
• Dosis:
– Forma crónica neuronopática identificada: iniciar 120 U/kg/15 días. Si la patología neurológica progresa, pasar a 240 U/kg/15 días durante 6 meses como máximo. Si no se produce mejora, disminuir la dosis a un nivel que permita controlar los síntomas viscerales.
– Niños en riesgo para desarrollar forma crónica neuronopática: iniciar 60 U/kg/15 días y vigilar de forma cuidadosa la evolución por si aparecen síntomas de alteración neurológica orgánica o funcional. Vigilar especialmente la normalidad de los movimientos sacádicos.
Tratamiento de la forma aguda neuronopática
• Pacientes tributarios de tratamiento:
– Puede ensayarse el tratamiento específico de estos pacientes con el objetivo de mejorar su calidad de vida. A efectos prácticos, es conveniente diferenciar entre los que tienen afectación piramidal y los que no la tienen.
• Tipo de tratamiento:
– Está autorizado el uso del TES y no está contemplado por el momento otro tipo de tratamientos.
• Dosis:
– Pacientes sin afectación piramidal y predominio de patología bulbar (estridor, dificultad para deglución): probar con 120 U/k cada 2 semanas; revisar dosis y la continuidad del tratamiento a los 6 meses de su inicio.
– Pacientes con afectación piramidal (opistótonos, espasticidad, trismo) y afectación cognitiva importante: ensayar una dosis de 15 U/k cada 2 semanas solamente para mejorar la visceromegalia.
Comentarios
El tratamiento enzimático sustitutivo para niños con formas no neuronopáticas de la enfermedad suele iniciarse, por lo general, con 60 unidades por kilo cada 15 días.
En los niños con enfermedad neuropática, especialmente en las formas agudas, no debe insistirse de forma indefinida en el tratamiento y es conveniente decidir conjuntamente con la familia la retirada del TES después de un tiempo prudencial (entre 6 y 12 meses) sin resultados terapéuticos satisfactorios.
Criterios de pérdida en el mantenimiento de los objetivos previamente alcanzados
Una vez instaurado el tratamiento y alcanzados los objetivos deseados, es necesario controlar si, como consecuencia de cambios en el tratamiento o de cualquier otro motivo, se produce una pérdida en el mantenimiento de los objetivos alcanzados.
• Criterios determinantes de pérdida:
– Si la Hb desciende 1,5 g/dL por debajo del valor previo a la reducción de la dosis.
– Si las plaquetas descienden un 25% por debajo del valor previo a la reducción de la dosis, o si la cifra es inferior a 80.000/mm3.
– Si aparece sangrado «espontáneo».
– Si el hígado o el bazo aumentan un 20% de volumen respecto al anterior.
– Si la enfermedad ósea progresa (empeora el dolor, fractura, infarto, necrosis).
– Si empeora la calidad de vida.
– Agravamiento de síntomas pulmonares, si los hay.
– Disminución del crecimiento.
• Criterios optativos:
– Aumento de quitotriosidasa (valorar variaciones que sean superiores al 5%).
– Descenso de la densidad mineral ósea.
Comentarios
En los pacientes no neuronopáticos, cuando se produce una pérdida de objetivos por disminución de la dosis, es necesario retomar la dosis con la que se habían obtenido y mantenido hasta ese momento los objetivos terapéuticos. Cuando la pérdida se produce sin disminución previa de la dosis, es necesario descartar la presencia de otra enfermedad concurrente causante del empeoramiento de los síntomas para proceder a su tratamiento, y si no se detecta esta última situación, es necesario valorar el aumento de la dosis.
En los niños con patología neurológica no existen parámetros cuantitativos definidos para pérdida de objetivos y debe ser considerada como tal cualquier agravamiento de la sintomatología que presentaban hasta ese momento.
Seguimiento
El seguimiento de los niños con enfermedad de Gaucher debe ser individualizado, pero el uso de un protocolo de «mínimos» facilita el control adecuado de los pacientes a largo plazo.
El seguimiento de los niños sin afectación neurológica debe sistematizarse en función de que estén sometidos o no al TES. En el primer caso, es preciso considerar si se ha obtenido una buena respuesta al tratamiento o todavía no se han alcanzado los objetivos terapéuticos diseñados en cada caso.
En los niños con enfermedad neurológica debe practicarse un seguimiento específico de esta patología, con independencia de los controles que puedan ser comunes con los otros pacientes y de acuerdo con las exigencias derivadas de su propia situación clínica.
Algoritmo para el tratamiento por objetivos de la enfermedad de Gaucher en la infancia
Con las recomendaciones recogidas en esta guía es posible diseñar un algoritmo para el tratamiento individualizado de cada paciente, que es el objetivo final deseado en el tratamiento de los niños con enfermedad de Gaucher.
Bibliografía
Barton NW, Brady RO, Dambrosia JM, Di Bisceglie AM, Dopplet SH, Hill SC, et al. Replacement therapy for inherited enzyme deficiency-macrophage targeted glucocerebrosidase for Gaucher´s disease. N Engl J Med. 1991; 324: 1.464-1.470.
Stocker JT, Dehner LP. Pediatric phatology. Filadelfia: JB Lippincott, 1992.
Downey MT. Estimation of splenic weight from ultrasonographic measurements. Can Assoc Radiol J. 1992; 43: 273-277.
Vellodi A, Bembi B, de Villemeur TB, Collin-Histed T, Erikson A, Mengel E, et al. Neuronopathic Gaucher disease task force of the European Working Group on Gaucher Disease. Management of neuronopathic Gaucher disease: A European consensus. J Inherit Metab Dis. 2001; 24: 319-327.
Cox TM, Aerts FG, Andria G, Beck M, et al. The Advisory Council to the European Working Group on Gaucher Disease (EWGGD). The role of the iminosugar N-butyldeoxynojirimicyn (Miglustat) in the management of the type I (non neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis. 2003; 26: 513-526.
Kluhs L, Teichgraber UK, Schneider U, Ludwing WD, Dorken B, Benter T. Accuracy of the sonographic determination of the splenic weight in comparison with the weight at autopsy. Fortschr Röntgenster. 2003; 175: 532-535.
Grabowski GA. Gaucher disease lessons from a decade of therapy. J Pediatr. 2004; 144: S15-S19.
Charrow J, Andersson HC, Kaplan P, Kolodny EH, Mistry P, Pastores G, et al. Enzyme replacement therapy and monitoring for children with type 1 Gaucher disease: consensus recommendations. J Pediatr. 2004; 144: 112-119.
Baldellou A, Andrea G, Campbell PE, Charrow J, Cohen IJ, Grabowski GA, et al. Paediatric non-neuropathic Gaucher disease: recommendations for treatment and monitoring. Eur J Pediatr. 2004; 163: 67-75.
Lebel E, Dweck A, Foldes J, Golowa Y, Itzchaky M, Zimran A, et al. Bone changes with enzyme therapy for Gaucher disease. J Bone Miner Metab. 2004; 22: 597-601.
Grabowski GA. Recent clinical progress in Gaucher disease. Curr Opin Pediatr. 2005; 17: 519-524.
Andersson HC, Charrow J, Kaplan P, Midtry P, Pastores GM, Prakash-Cheng A, et al. Individualization of long term enzyme replacement therapy for Gaucher disease. Genet Med. 2005; 7(2): 105-110.
Weinberg NJ, Barranger JA, Charrow J, Grabowski GA, Mankin HJ, Mistry P. Guidance on the use of miglustat for treating patients with type 1 Gaucher disease. Am J Hematol. 2005; 80: 223-229.
Alentado N, Escrig R, Dalmau J. Enfermedad de Gaucher tipo I: 10 años de experiencia en el tratamiento enzimático sustitutivo. Acta Pediatr Esp. 2005; 63: 373-376.
Andrea G, Parenti G. Oligosaccharidosis and related disorders. En: Blau N, Hoffmann GF, Leonard J, Clarke JTR, eds. Physician´s guide to the treatment and follow-up of metabolic diseases. Berlín: Springer-Verlag, 2006; 205-215.
Lesiones faciales acneiformes en niña de 5 años
La esclerosis tuberosa es una enfermedad neurocutánea de herencia autosómica dominante y expresividad variable que afecta a la diferenciación y proliferación celular con la formación de hamartomas en distintos órganos. A pesar de que se han identificado dos genes, TSC1 y TSC2, situados en los cromosomas 9:9q34 y 16:16p13, respectivamente, el diagnóstico de la enfermedad es clínico ya que los tests genéticos tienen un porcentaje elevado de falsos negativos (15%). Las manifestaciones clínicas varían enormemente, incluso entre familiares. La presencia de lesiones cutáneas características es de gran utilidad para orientar el diagnóstico. El enfoque debe ser multidisciplinario ya que puede existir afectación renal, pulmonar, cardiaca o del sistema nervioso central con importante morbilidad. Es importante prestar atención a dichas complicaciones sistémicas para minimizar las secuelas de la esclerosis tuberosa.
Secuencia de Poland: aportación de dos nuevos casos
La secuencia de Poland es una anomalía musculosquelética congénita caracterizada por la ausencia de la porción esternal del músculo pectoral mayor asociada a braquisindactilia. Se presenta con una incidencia aproximada de 1/20.000-30.000 nacidos vivos (un 75% de los casos corresponde a varones), y en un 70% está afectado el lado derecho. Además de la asociación con la braquisandactilia, la ausencia del pectoral mayor puede presentarse aislada o bien asociada a otras malformaciones, especialmente de la zona pectoral o del brazo y la mano homolaterales, como sindactilia, ausencia del pectoral menor, hipoplasia de la extremidad o amastia. También se puede asociar a alteraciones de otros territorios, como dextrocardia, síndrome de Moebius o neurofibromatosis.
La etiología es desconocida y habitualmente no presenta base hereditaria. Se ha propuesto como teoría patogénica una hipoplasia de la arteria subclavia fetal. El tratamiento es quirúrgico, fundamentalmente con fines estéticos o para corregir las alteraciones funcionales de la mano que esta anomalía conlleva.
Aportamos dos casos tratados en nuestro servicio de pediatría: uno corresponde a un varón recién nacido, con afectación del pectoral mayor e hipoplasia del arco anterior de las costillas 2-5, y el otro a una niña de 9 años afectada de asma mediada por inmunoglobulina E (IgE), que asocia a la agenesia del pectoral mayor una hipoplasia de la mama, una hipoplasia de la mano y braquisindactilia.
Tabique vaginal transverso completo como causa de hematocolpos y dolor abdominal agudo
Sr. Director:
El tabique vaginal transverso es una malformación extremadamente rara. Se debe a un defecto embrionario que puede permanecer oculto si el tabique es incompleto y requiere intervención quirúrgica si es completo. Exponemos el caso clínico de una niña con esta anomalía que presentó dolor abdominal agudo debido al hematocolpos que el tabique había producido, como consecuencia de la obstrucción de la salida del flujo menstrual.
Se trata de una niña de 12 años de edad, de procedencia sudamericana, sin menarquia, que acudió al servicio de urgencias por presentar un dolor abdominal intenso de 72 horas de evolución, de tipo cólico, localizado en el hipogastrio. No presentaba fiebre, vómitos ni sintomatología urinaria. El hábito dietético era normal, así como las deposiciones. En la exploración se detectaba un buen estado general y nutricional. Mostraba un desarrollo mamario y vello pubiano en estadio III de Tanner. El abdomen era blando y depresible, sin visceromegalias. Presentaba un discreto dolor a la palpación profunda en el hipogastrio y la región suprapúbica, sin masas palpables. Los puntos apendiculares eran negativos y no había signos de irritación peritoneal. Los genitales externos eran normales.
Las pruebas analíticas practicadas en urgencias, que incluían hemograma, bioquímica, coagulación y orina simple, eran normales. En la ecografía abdominal se detectó una imagen bien delimitada y homogénea, de contenido hemático de 9 3 8 cm de diámetro, localizada en el interior de la vagina, que llegaba a desplazar a la vejiga. Ante la ausencia de un himen imperforado y la posibilidad de un hematocolpos debido a otra malformación, se ingresa a la paciente para su estudio y tratamiento. Se completó el estudio con nuevas ecografías de abdomen, que confirmaron el hallazgo descrito y no mostraron malformaciones en ninguna otra localización. La radiografía de tórax y el estudio cardiológico eran normales y los cultivos de orina estériles. Bajo control ecográfico se realizó el drenaje de 430 mL de contenido hemático y se visualizó un tabique vaginal transverso completo en el tercio medio-inferior de la vagina de 1,5 cm de espesor. Se practicó una resección completa del tabique y, posteriormente, aparecieron menstruaciones normales.
La presencia de un tabique vaginal transverso es una de las patologías más raras del tracto reproductivo y se debe a una alteración de la fusión vertical de los conductos de Müller1. Fue descrito por primera vez por Delanuy, en 1877, y desde entonces se han publicado muy pocos casos. Cuando el tabique es completo (imperforado), se produce, con las primeras menstruaciones, un hematocolpos, y como consecuencia de ello, un cuadro obstructivo, como en este caso. Cuando es incompleto (perforado), el drenaje menstrual puede ser normal, pero puede ponerse de manifiesto al mantener relaciones sexuales.
La prevalencia de esta anomalía se estima en 1/72.000 pacientes ginecológicas; incluso es probable que esta alteración sea menos frecuente que la ausencia congénita de vagina y útero2. Suidan y Azoury aportan 12 casos de tabique vaginal transverso en mujeres de 16-36 años de edad y sólo uno de ellos, en una paciente de 16 años, era completo3. Una mínima parte de casos de tabique vaginal transverso presenta otras malformaciones asociadas con el aparato reproductor, como atresia vaginal, malformaciones de útero, atresia o agenesia de trompas de Falopio y agenesia de ovario. También se han descrito malformaciones renales (ectopia de uréteres), cardiacas (coartación de aorta y defectos del septum interauricular) y del aparato digestivo (malrotación intestinal y ano imperforado)4-6. Incluso se han publicado casos asociados a himen imperforado7,8. En más del 40% de los casos, la localización del tabique se encuentra en el tercio superior de la vagina9.
Clínicamente, se manifiesta en las niñas mayores con amenorrea primaria y con signos y síntomas compresivos producidos por el hematocolpos, como dolor, megauréter, hidronefrosis o estreñimiento, e incluso puede producir una compresión de la vena cava inferior y un edema en las extremidades inferiores. En cualquier caso, se puede distinguir entre un tabique transverso y un himen imperforado por la ausencia de distensión en el introito vaginal con la maniobra de Valsalva. La ecografía ginecológica puede ser suficiente para el diagnóstico, aunque la ecografía transrectal es la prueba más adecuada para un diagnóstico correcto. La resección del tabique es efectiva si no se presentan complicaciones, y la evolución es favorable en la mayor parte de las pacientes10.
La presentación de este caso tiene por objeto considerar la presencia de este defecto embrionario ante un cuadro agudo de dolor abdominal hipogástrico en una niña con hematocolpos y también ante una niña con desarrollo puberal completo, amenorrea primaria y síntomas de obstrucción intestinal.
Bibliografía
- Folch M, Pigem I, Monje JC. Mullerian agenesis: etiology, diagnosis and management. Obst Ginecol Surg. 2000; 56: 644-649.
- Martínez M, Pérez I, Tineo Z, Rodríguez Y. Tabique vaginal transverso completo. Caso clínico. Informed. 2007; 9: 267-271.
- Suidan FG, Azoury RS. The transverse vaginal septum: a clinicopathology evaluation. Obstet Gynecol. 1979; 54: 278-283.
- Bustos P, Smirnow M. Tabique vaginal transverso y atresia vaginal. Rev Chil Obstet Ginecol. 2003; 68: 229-234.
- Chin AI, Rutman M, Razs. Transverse vaginal septum with congenital vesical-vaginal communication and cyclical hematuria. Urology. 2007; 69: 575.
- Nazir Z, Rizvi RM, Qureshi RN, Khan ZS, Khan Z. Congenital vaginal obstructions: varied presentation and outcome. Pediatr Surg Int. 2006; 22: 749-753.
- Deligeoroglou E, Deliveliotou A, Makrakis E, Creatsas G. Concurrent imperforate hymen, transverse vaginal septum, and unicornate uterus: a case report. J Pediatr Surg. 2007; 42: 1.446-1.448.
- Ahmed S, Morris LL, Atkinson E. Distal mucocolpos and proximal hematocolpos secondary to concurret imperforate hymen and transversal vaginal septum. J Pediatr Surg. 1999; 34: 1.555-1.556.
- Wenof M, Reyniak JV, Novendster J, Castadot MJ. Transverse vaginal septum. Obstet Gynecol. 1979; 54: 60-63.
- Rana A, Manandhar B, Amatya A, Baral J, Gurung G, Giri A, et al. Mucocolpos due to complete transverse septum in middle third of vagina in a 17-year-old girl. J Obstet Gynaecol Res. 2002; 28: 86-88.
Aplasia cutánea congénita
La aplasia cutánea congénita (ACC) es una rara alteración caracterizada por la ausencia congénita de epidermis, dermis y, en ocasiones, de los tejidos subyacentes. La forma más frecuente afecta al vértex, y se puede presentar de forma aislada o asociada a otras malformaciones. Fue descrita por primera vez por Cordon, en 1767, y desde entonces se han documentado más de 500 casos. La frecuencia se ha estimado en torno a 3/10.000 recién nacidos. No hay predilección por la raza ni el sexo. La etiopatogenia no está clara. La manifestación clínica habitual es una lesión oval o circular, solitaria, sin pelo, bien delimitada, de 0,5-2 cm, y localizada en el vértex. Al nacer, las lesiones pueden estar cicatrizadas o presentarse como erosiones o incluso úlceras profundas, con riesgo de afectar a las meninges o la duramadre. La mejor opción terapéutica dependerá del tamaño del defecto.
Síndrome de Moebius: diagnóstico neonatal
El síndrome de Moebius es un trastorno congénito caracterizado por la asociación de parálisis facial y afectación de la musculatura oculomotora secundaria a la agenesia de los pares craneales VII y VI. Pueden estar implicados otros pares craneales y/o asociarse otras malformaciones como consecuencia de una disrupción en la morfogénesis troncoencefálica durante el periodo embrionario, presumiblemente de carácter vascular. Se presenta un caso de síndrome de Moebius diagnosticado durante el periodo neonatal, realizando una revisión de sus características clínicas y evolutivas.
Asfixia perinatal: relación entre afectación cardiovascular, neurológica y multisistémica
Objetivo: Conocer el perfil epidemiológico de los neonatos a término con asfixia perinatal; relacionarlo con la existencia y el grado de encefalopatía hipóxico-isquémica (EHI); valorar la frecuencia de afectación cardiovascular (ACV) y relacionarla con la afectación neurológica y extraneurológica; determinar la relación entre ACV y factores etiopatogénicos de la asfixia.
Material y métodos: Estudio retrospectivo de los pacientes que cumplieron criterios de asfixia perinatal entre enero de 2000 y diciembre de 2004.
Resultados: Se incluyeron 295 pacientes. Un 39% cumplía criterios de EHI: leve 23,1%; moderada 8,8% y grave 7,1%. La afectación pulmonar se dio en un 35,9%, la renal en un 18%, presentaron hipocalcemia un 18,6%, trombopenia un 13,9% y coagulopatía un 21,4%. Un 14,2% de los pacientes presentaron ACV cierta (alteración enzimática y/o ecocardiográfica con shock y/o hipotensión arterial), y un 15,6% ACV probable (sólo hipotensión arterial y/o shock). La existencia de ACV se relaciona con la presencia de EHI y, por consiguiente, con alteraciones en el electroencefalograma y de neuroimagen. Asimismo, la existencia de ACV aumenta la presencia de afectación extraneurológica. También se constata que los pacientes con ACV presentan más frecuentemente acidosis al ingreso y acidosis metabólica persistente durante su evolución.
Conclusiones: La ACV se correlaciona con la existencia y la gravedad de las manifestaciones neurológicas y con la afectación de otros órganos y sistemas, en especial con la acidosis metabólica persistente.
Tumor de Wilms en paciente trasplantado renal
Los pacientes portadores de trasplantes tienen un riesgo superior al resto de la población de padecer neoplasias. En el caso de los trasplantados renales éste es aún mayor, pues, además de tomar una medicación inmunodepresora, en muchas ocasiones han estado expuestos durante largos periodos de tiempo a técnicas de diálisis, con el consiguiente riesgo de haber desarrollado enfermedad renal quística adquirida. Se expone el caso de una niña de 6 años portadora de un trasplante renal normofuncionante desde los 2 años, que acude a urgencias por presentar un cuadro de dolor abdominal, diagnosticada finalmente de un tumor de Wilms en el riñón nativo izquierdo. Se incide sobre la utilidad de la ecografía en este tipo de pacientes (hay protocolos que recomiendan su realización periódica como método de cribado de procesos neoplásicos) y sobre la importancia de reconocer determinados síndromes o condiciones predisponentes al desarrollo de tumores en este grupo de pacientes, que modificarán la actitud terapéutica.
Las cepas enterohemorrágicas de «Escherichia coli» como paradigma de patógenos emergentes. Lecciones de la gran epidemia de infección alimentaria centrada en Alemania en mayo y junio de 2011
Las cepas enterohemorrágicas de Escherichia coli (EHEC) son patógenos zoonóticos de transmisión alimentaria asociados a epidemias y a casos esporádicos de diarrea y colitis hemorrágica, cuadros que pueden complicarse y dar lugar al síndrome hemolítico urémico (HUS). La importancia de las cepas EHEC se debe a la gravedad del HUS, que es la causa más frecuente de insuficiencia renal aguda en los niños en América y Europa. E. coli O157:H7 es el serotipo fundamental del patotipo EHEC. Ya hace años se predijo que cepas EHEC de otros serotipos podrían convertirse en importantes patógenos transmitidos por alimentos, y desde entonces estos microorganismos se han relacionado con numerosos casos de enfermedad en todo el mundo. La incidencia de estos serotipos distintos del O157:H7 sigue creciendo, lo que significa que pueden considerarse patógenos emergentes. Un ejemplo reciente es el gran brote de infecciones transmitidas por los alimentos y causadas por EHEC O104:H4, que se centró principalmente en Alemania durante mayo y junio de 2011. La cepa responsable del brote posee una combinación de factores de virulencia típicos de diferentes patotipos de E. coli, lo que corrobora que la plasticidad de los genomas bacterianos facilita la emergencia de nuevos patógenos especialmente virulentos. Las investigaciones epidemiológicas identificaron el origen del brote en una partida de semillas de alholva (fenogreco) importadas de Egipto en 2009. La dimensión internacional de esta epidemia ilustra la urgente necesidad de mejorar la vigilancia epidemiológica de las cepas EHEC.
Síndrome de Rud: observación de un nuevo caso
La asociación de la tríada oligofrenia, ictiosis congénita e hipogonadismo constituye el síndrome de Rud, entidad con muy pocos casos en la literatura médica mundial. Se han descrito en estos pacientes otras alteraciones menos frecuentes, como crisis convulsivas, epilepsia, talla baja, retinitis pigmentosa, polineuropatía hipertrófica o sordera neurosensorial. Forma parte de los denominados síndromes neurocutáneos queratósicos, junto con otros mejor definidos, como el síndrome de Sjögren-Larsson o la enfermedad de Refsum. Se expone el caso de un paciente de 13 años de edad, con criterios clínicos compatibles con el síndrome de Rud, que asocia una agenesia renal unilateral y un sarcoma de partes blandas.
Úlceras acrales y fenómeno de Raynaud en un adolescente
El síndrome de Crest es una variedad de esclerodermia sistémica; concretamente, una forma limitada. Se considera una forma incompleta de la enfermedad, que presenta un curso más benigno, dado que las afectaciones renal y pulmonar no son sus principales características.
Los síntomas fundamentales del síndrome de Crest, que conforman su acrónimo, son: calcinosis, fenómeno de Raynaud, dismotilidad esofágica, esclerodactilia y telangiectasias.
Presentamos un caso cuyo inicio se produjo durante la adolescencia, hecho muy poco frecuente, que, sin embargo, presenta un mejor pronóstico que en la edad adulta.
Rehidratación intravenosa rápida en urgencias: comparación con la pauta tradicional
Objetivo: Determinar si la rehidratación rápida es un método efectivo para corregir la deshidratación en niños con gastroenteritis aguda (GEA) y deshidratación leve-moderada, y comprobar si permite restablecer en menos tiempo el estado de hidratación, en comparación con el método tradicional.
Métodos: Se trata de un estudio retrospectivo, analítico-descriptivo, llevado a cabo en niños atendidos en un servicio de urgencias, con una deshidratación leve-moderada isonatrémica, desencadenada por una GEA. Se recopilaron un total de 42 casos y se compararon dos pautas de rehidratación: rápida (19 casos) y tradicional (23 casos).
Resultados: El tiempo empleado para la rehidratación i.v. fue menor con la pauta rápida que con la tradicional, con una media de 3,53 horas (desviación estándar [DE]= 1,3) y 12,04 horas (DE= 4,08), respectivamente. Esta diferencia fue estadísticamente significativa (p <0,01). También se encontraron diferencias estadísticamente significativas en cuanto al tiempo necesario para alcanzar una tolerancia oral exitosa, con una media de 6,37 horas (DE= 3,63) en el grupo de la pauta rápida, frente a 9,35 horas (DE= 5,16) en el de la pauta tradicional (p= 0,04). Por el contrario, a pesar de que el tiempo medio de estancia en el servicio de urgencias fue menor para la pauta rápida y que la ganancia de peso fue mayor que en la pauta tradicional, estas diferencias no fueron estadísticamente significativas.
Conclusión: La rehidratación i.v. rápida supone una alternativa a la pauta clásica con una serie de ventajas, como la facilidad de cálculo con menor posibilidad de errores, una mejora más rápida del estado de hidratación y del estado general, que permite una tolerancia oral más precoz, o la corrección más rápida de las alteraciones electrolíticas y del equilibrio ácido-base gracias a la restauración precoz de la perfusión renal, y todo ello de forma segura, sin que aumente el riesgo de complicaciones como la hipernatremia.
Síndrome de apnea obstructiva del sueño con repercusión sistémica. A propósito de un caso
Presentamos el caso de un niño de 4 años que ingresa en la unidad de cuidados intensivos pediátricos (UCIP) por mal estado general, dificultad respiratoria severa con débil esfuerzo y ronquido inspiratorio. La auscultación cardiopulmonar muestra hipoventilación y ritmo de galope, tiene hepatomegalia y, desde el punto de vista neurológico, presenta disminución del nivel de conciencia (Glasgow 6/15). En las pruebas complementarias, se observa un daño hipóxico-isquémico generalizado (creatinina sérica de 1,8 mg/dL; GOT de 23.730 UI/L y GPT de 5.771 UI/L; actividad de protrombina del 31% y troponina de 1,73 ng/mL). La radiografía de tórax muestra una discreta cardiomegalia y la ecocardiografía hipertensión pulmonar. En la eco-Doppler abdominal se observa una severa hiperecogenicidad cortical renal y hepatomegalia. En el electroencefalograma hay signos de afectación cerebral generalizada, y en la tomografía computarizada (TC) craneal aparecen dos áreas cerebrales sugestivas de infartos isquémicos e hipertrofia adenoidea. A los pocos días del ingreso, se realiza una adenoamigdalectomía. En el momento del alta, los parámetros analíticos son normales, y en la ecocardiografía no se observa hipertensión pulmonar.
Diagnóstico y manejo del absceso hepático amebiano
Sr. Director:
Debido a los movimientos migratorios, cada vez es más común encontrarse en nuestro medio con enfermedades propias de climas tropicales. Describimos un caso en el que coexisten varias enfermedades tropicales importadas, entre ellas el absceso hepático amebiano.
Se trata de una niña de 9 años de edad, procedente de Guinea Ecuatorial, que reside en España desde hace un mes. Consulta por fiebre y dolor en el hipocondrio derecho de 3 días de evolución. Presenta hepatomegalia dolorosa y esplenomegalia. Las exploraciones complementarias pusieron de manifiesto los siguientes resultados: anemia microcítica hipocrómica, leucocitosis moderada sin desviación izquierda, eosinofilia moderada, ferropenia, proteína C reactiva 35 mg/dL, velocidad de sedimentación globular 108 mm/h, bilirrubina normal, transaminasas normales, fosfatasa alcalina y gamma-glutamil transferasa ligeramente elevadas. Gota gruesa: Plasmodium falciparum (parasitemia baja, <1%). VIH negativo. Serologías de hepatitis A, B y C negativas.
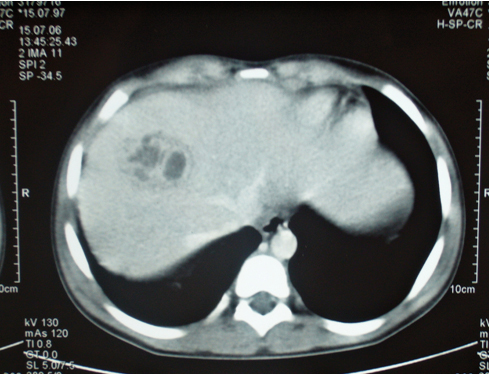
A pesar del tratamiento correcto de la malaria (quinina y doxiciclina orales, ante la procedencia de una zona endémica de malaria resistente a cloroquina) y la negativización de la parasitemia, la paciente continúa presentando picos de fiebre alta, dolor abdominal y vómitos. Se realiza una ecografía abdominal, que muestra una imagen sugestiva de abscesos, por lo que se solicita una tomografía computarizada abdominal con contraste oral e intravenoso, en la que se visualizan varios abscesos hepáticos (figura 1) y multitud de gusanos en la luz intestinal (figura 2). Se instaura tratamiento intravenoso con cefotaxima y metronidazol. A posteriori se confirma la elevación de anticuerpos anti-Entamoeba histolytica (1/2560). Asimismo, se realiza un tratamiento con mebendazol oral durante 3 días ante la presencia de parásitos en el frotis de heces (Ascaris lumbricoides, Trichuris trichiura y oxiuros). La evolución es satisfactoria, y un año después no se han detectado restos del absceso hepático en el control ecográfico.
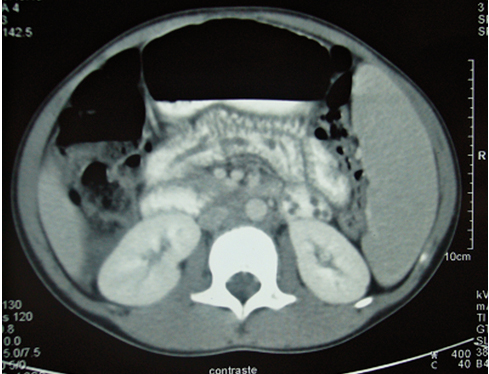
E. histolytica es un agente infeccioso que puede detectarse en todo el mundo, con una prevalencia de hasta el 50% en los países en vías de desarrollo. El síndrome clínico más frecuente es la infección no invasiva intestinal. En otras ocasiones produce disentería con diarrea sanguinolenta o amebomas. Las localizaciones extraintestinales incluyen los abscesos hepáticos y, menos frecuentemente, afectación pulmonar, cardiaca y cerebral1. Los trofozoítos invaden la mucosa del colon y alcanzan el hígado a través de la circulación portal. La clínica puede comenzar días o meses después de una disentería, pero también puede aparecer sin antecedentes de amebiasis intestinal. Consiste en fiebre, decaimiento, náuseas, vómitos, dolor abdominal irradiado a la espalda o al hombro derecho, hepatomegalia dolorosa y tos improductiva. La ictericia y la diarrea no son frecuentes2. Dado que los resultados analíticos son inespecíficos, el diagnóstico inicial es ecográfico. En las formas extraintestinales el examen de las heces puede ser negativo, por lo que son necesaria las pruebas serológicas. La técnica IHA (indirect hemagglutination antibody) es la más sensible (90-100%) en la amebiasis hepática. El diagnóstico definitivo consiste en aislar E. histolytica en el material biopsiado3,4. El tratamiento se debe realizar con metronidazol intravenoso y un amebicida intraluminal (paromomicina). No suele ser necesario el drenaje quirúrgico o mediante punción-aspiración percutánea guiada por ecografía, excepto en casos resistentes al tratamiento médico, contraindicaciones del metronidazol (embarazo) o en presencia de signos de inminente rotura o extensión pleuropulmonar5. La complicación más frecuente es la rotura del absceso y su extensión a la pleura, el pericardio y/o el peritoneo, lo que conlleva una alta mortalidad6.
Bibliografía
- Pritt BS, Clark CG. Amebiasis. Mayo Clin Proc. 2008; 83: 1.154-1.160.
- Salles JM, Moraes LA, Salles MC. Hepatic amebiasis. Braz J Infect Dis. 2003; 7: 96-110.
- Salles JM, Salles MJ, Moraes LA, Silva MC. Invasive amebiasis: an update on diagnosis and management. Exp Rev Anti-Infect Ther. 2007; 5: 893-901.
- Yost J. Amebiasis. Pediatr Rev. 2002; 23: 293-294.
- Blessmann J, Binh HD, Hung DM, Tannich E, Burchard G. Treatment of amoebic liver abscess with metronidazole alone or in combination with ultrasound-guided needle aspiration: a comparative, prospective and randomized study. Trop Med Int Health. 2003; 8: 1.030-1.034.
- Rao S, Solaymani-Mohammadi S, Petri WA Jr, Parker SK. Hepatic amebiasis: a reminder of the complications. Curr Opin Pediatr. 2009; 21: 145-149.
Resultados de una serie de biopsias renales percutáneas guiadas ecográficamente en una población pediátrica: nuestra experiencia en 15 años
Objetivos: Presentar nuestra experiencia sobre biopsias renales percutáneas guiadas ecográficamente en pacientes en edad pediátrica desde que se instauró dicha técnica en nuestro hospital, y valorar la correlación clínica/anatomopatológica y las complicaciones de la misma.
Pacientes y métodos: Estudio retrospectivo de 180 biopsias renales percutáneas guiadas ecográficamente realizadas a un total de 164 pacientes (de 6 meses-18 años de edad) durante un periodo de 15 años. El protocolo de la biopsia incluye su realización en el quirófano mediante sedación y una ecografía a las 24 horas.
Resultados: El motivo más frecuente de su realización fue la presencia de un síndrome nefrótico corticodependiente/resistente (29,4%), seguido de la proteinuria de diverso rango con presencia de hematuria. El diagnóstico anatomopatológico más habitual fue la glomerulonefritis mesangial por IgA (26,1%), seguido de cambios glomerulares mínimos, confirmándose en la mayoría de los casos la sospecha clínica. Únicamente se detectó una complicación grave (hematoma renal/hipotensión arterial) en un paciente de riesgo.
Conclusiones: En nuestra experiencia, la biopsia renal percutánea es un método diagnóstico fiable y seguro, independientemente de la edad
Pezón supernumerario: ¿un marcador cutáneo de anomalías hematológicas, cardiovasculares y renales?
Sr. Director:
 Los pezones supernumerarios (PS), también denominados pezones accesorios o politelia, son una anomalía congénita menor, relativamente común, y constituyen la patología mamaria accesoria más frecuente (tabla 1). Representan restos de las crestas mamarias embriológicas, engrosamientos ectodérmicos simétricos que se extienden desde la axila hasta la ingle. Para algunos autores, son un ejemplo de atavismo o aparición espontánea de características ancestrales en los miembros de una especie. Antiguamente se asociaban con la fertilidad, y en la época medieval se consideraron una marca del diablo1.
Los pezones supernumerarios (PS), también denominados pezones accesorios o politelia, son una anomalía congénita menor, relativamente común, y constituyen la patología mamaria accesoria más frecuente (tabla 1). Representan restos de las crestas mamarias embriológicas, engrosamientos ectodérmicos simétricos que se extienden desde la axila hasta la ingle. Para algunos autores, son un ejemplo de atavismo o aparición espontánea de características ancestrales en los miembros de una especie. Antiguamente se asociaban con la fertilidad, y en la época medieval se consideraron una marca del diablo1.
Según las series, su prevalencia varía desde el 0,22 hasta el 6%, son más frecuentes en los sujetos de raza negra, asiáticos, indígenas americanos, árabes y judíos que en los europeos de raza caucásica. No hay diferencias según el sexo, aunque se detecta un ligero predominio en los varones. Normalmente es una anomalía esporádica, aunque el 6-10% de los casos son familiares (herencia autosómica dominante con penetrancia incompleta o dominante ligada al cromosoma X). Los PS se localizan habitualmente en la región inframamaria, sobre todo la izquierda (figura 1). Pueden presentarse en la región supramamaria o en cualquier otra zona de las líneas mamarias embriológicas. En ocasiones se sitúan fuera de esas líneas, como la espalda, los hombros, la cara posterior de los muslos, la cara, el cuello o la vulva. Las lesiones suelen ser solitarias, pero hay casos múltiples (incluso 8) unilaterales o bilaterales. Clínicamente, se manifiestan como tumores pediculados pequeños, blandos y rosados o marrones. En el recién nacido las lesiones pueden ser muy tenues, en forma de máculas de 1-3 mm de tamaño y de color marrón claro2.
Generalmente, el diagnóstico se basa en la clínica y en su presencia desde el nacimiento. Pueden confirmarse mediante dermatoscopia, nuevas técnicas, como la microscopia confocal de reflexión, o el estudio histopatológico. Éste muestra el engrosamiento epidérmico, las estructuras pilosebáceas y el músculo liso, con o sin glándulas mamarias verdaderas. El diagnóstico diferencial se realiza con el nevo melanocítico, el neurofibroma, las cicatrices de amniocentesis y el pólipo anexial neonatal. Los PS son un proceso benigno, pero pueden desarrollar cualquier enfermedad que aparezca en las mamas normales, incluidos los tumores benignos y malignos. Normalmente no precisa tratamiento, pero en casos sintomáticos, o por motivos estéticos, pueden extirparse1,3.
Los PS se asocian a varios síndromes polimalformativos, como el síndrome de Simpson-Golabi-Behmel (trastorno de herencia recesiva ligada al cromosoma X caracterizado por sobrecrecimiento prenatal y posnatal, alteraciones craneofaciales, anomalías congénitas cardiacas, renales y/o esqueléticas y tumores embrionarios), varios subgrupos del síndrome de displasia ectodérmica, el síndrome 3-M, el síndrome de Killian/Teschler-Nicola y la disostosis espondilocostal. También se ha observado una coexistencia con el nevo de Becker o la lentiginosis unilateral parcial1.
Lo más importante de los PS es su posible relación con algunas enfermedades y, por tanto, su posible utilidad como marcador cutáneo. Aunque esta relación es controvertida4, recientemente han aparecido nuevos estudios que la apoyan5-7. Entre dichas enfermedades se incluyen las hematológicas (deficiencias de factores y leucemias agudas)5, las cardiovasculares6,8, el cáncer renal o genital9 y las malformaciones renales y de las vías urinarias7,10. Algunos autores explican esta última asociación por un desarrollo embriológico paralelo del sistema genitourinario y mamario. Mientras que algunos autores recomiendan realizar en todos los pacientes con PS7 un estudio renal, especialmente mediante ecografía, otros lo reservan para los casos familiares10 o los asociados a otras malformaciones3.
Bibliografía
- Antaya R, Schaffer JV. Anomalías del desarrollo. En: Bolognia JL, Jorizzo JL, Rapini RP, eds. Dermatología, 1.ª ed. Madrid: Elsevier España, S.A.; 2004; 915-931.
- Brown J, Schwartz RA. Supernumerary nipples: an overview. Cutis. 2003; 71: 344-346.
- Merlob P. Congenital malformations and developmental changes of the breast: a neonatological view. J Pediatr Endocrinol Metab. 2003; 16: 471-485.
- Grotto I, Browner-Elhanan K, Mimouni D, Varsano I, Cohen HA, Mimouni M. Occurrence of supernumerary nipples in children with kidney and urinary tract malformations. Pediatr Dermatol. 2001; 18: 291-294.
- Aslan D, Gürsel T, Kaya Z. Supernumerary nipples in children with hematologic disorders. Pediatr Hematol Oncol. 2004; 21: 461-463.
- Rajaratnam K, Kumar PD, Sahasranam KV. Supernumerary nipple as a cutaneous marker of mitral valve prolapse in Asian Indians. Am J Cardiol. 2000; 86: 695-697.
- Ferrara P, Giorgio V, Vitelli O, Gatto A, Romano V, Bufalo FD, et al. Polythelia: still a marker of urinary tract anomalies in children? Scand J Urol Nephrol. 2009; 43: 47-50.
- Urbani CE. Supernumerary nipple and cardiocutaneous associations. J Am Acad Dermatol. 2004; 50: e9.
- Urbani CE, Betti R. Aberrant mammary tissue and nephrourinary malignancy. Cancer Genet Cytogenet. 1996; 87: 88-89.
- Brown J, Schwartz RA. Supernumerary nipples and renal malformations: a family study. J Cutan Med Surg. 2004; 8: 170-172.
Síndromes de Pearson y de Kearns-Sayre: dos enfermedades mitocondriales multisistémicas, debidas a deleciones en el ADN mitocondrial
El síndrome de Pearson (SP) y el síndrome de Kearns-Sayre (SKS) son enfermedades mitocondriales multisistémicas con diferente fenotipo, causadas por deleciones en el ADN mitocondrial (ADNmt).
Objetivo: Describir las manifestaciones clínicas y los hallazgos neurorradiológicos, bioquímicos y genético-moleculares de ambos síndromes, con objeto de difundir su conocimiento entre los pediatras.
Pacientes y métodos: Se han estudiado retrospectivamente 6 pacientes con SKS y 3 con SP inicial, dos de los cuales evolucionaron a SKS.
Resultados: La edad de inicio de los síntomas fue inferior en los SP. Los síntomas más precoces fueron los hematológicos (anemia), seguidos de los renales (Fanconi) y digestivos (insuficiencia pancreática), y de forma más tardía se presenta la afectación ocular, endocrinológica, cardiológica y neurológica. Cuatro pacientes precisaron implantación de marcapasos. Seis casos presentaron alteraciones cerebrales y/o del tronco del encéfalo en la resonancia magnética. Se observó hiperlactatorraquia, hiperproteinorraquia y descenso de ácido fólico en el líquido cefalorraquídeo. La mitad de los SKS presentaron fibras musculares rojo-rasgadas y fibras citocromo C oxidasa negativas. En ocho pacientes se detectó una deleción única del ADNmt.
Conclusiones: 1) Las diferencias más acusadas entre el SP y el SKS fueron la edad de comienzo y las manifestaciones iniciales. Los síntomas en la evolución, así como los hallazgos bioquímicos, neurorradiológicos y genéticos, fueron similares. 2) Las enfermedades mitocondriales deberían incluirse en el diagnóstico diferencial del síndrome de Fanconi, el déficit de la hormona del crecimiento y los trastornos de la conducción cardiaca, especialmente en los casos con afectación multiorgánica. El diagnóstico se confirma por la presencia de una gran deleción en el ADNmt.
Necrosis grasa subcutánea en un recién nacido y sus complicaciones
Sr. Director:
La necrosis grasa subcutánea (NGS) del recién nacido es una paniculitis lobulillar transitoria1, poco frecuente en la infancia, de carácter autolimitado, que aparece en las primeras semanas de vida en neonatos a término y postérmino como consecuencia de situaciones de estrés en el periodo neonatal2. Es una entidad benigna, autorresolutiva en varias semanas, aunque puede ir asociada a complicaciones extracutáneas, como hipoglucemia, trombocitopenia, hipertrigliceridemia y, la más grave, hipercalcemia3,4, que obliga a establecer una estrecha vigilancia de estos pacientes durante 6 meses.
Se presenta el caso de una paciente con necrosis grasa del recién nacido e hipercalcemia secundaria grave. El objetivo de este artículo es comentar la clínica, el tratamiento y la evolución del cuadro.
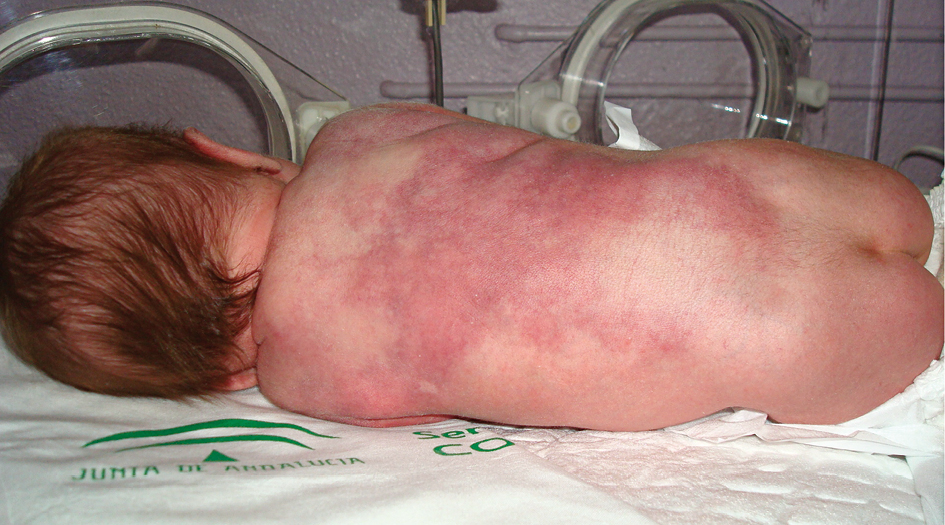
Neonato de sexo femenino, nacido a término y con un peso adecuado para la edad gestacional, con antecedentes de gestación controlada sin incidencias, parto por cesárea por pérdida del bienestar fetal y líquido meconial intraparto, por lo que precisó ventilación con bolsa y mascarilla (puntuación del test de Apgar 5/9). Los resultados de la exploración al nacimiento estaban dentro de la normalidad, salvo un soplo sistólico II/VI, que posteriormente fue diagnosticado de estenosis pulmonar leve. A las 48 horas de vida presentó una llamativa lesión eritematosa en la espalda (angioma), que a los 4 días había evolucionado a una gran placa eritematoso-violácea con nódulos calientes y dolorosos, extendiéndose de forma significativa a las regiones dorsal y lumbar, y a la raíz de los miembros superiores e inferiores (figura 1), acompañada de un síndrome febril. Ante el aumento de reactantes de fase aguda (proteína C reactiva 10,29 mg/dL), se extrajo un hemocultivo y se pautó antibioterapia empírica con ampicilina y gentamicina intravenosa, aislándose en el cultivo Staphylococcus epidermidis. En la ecografía abdominal no se visualizaba el riñón derecho, por lo que se realizó una gammagrafía renal DMSA que confirmó la agenesia renal derecha. Pocas horas después presentó un episodio de hiponatremia (119 mEq/L), edemas, proteinuria, oliguria y hematuria microscópica, que se diagnosticó como necrosis tubular aguda.
Tras el diagnóstico clínico de NGS, el servicio de dermatología realizó una biopsia de las lesiones que confirmó el diagnóstico. El estudio histopatológico mostraba una paniculitis lobulillar con infiltrado inflamatorio granulomatoso y presencia de hendiduras en forma de aguja, birrefringentes con luz polarizada y de disposición radial en el citoplasma de los adipocitos y de las células gigantes multinucleadas. Durante su ingreso las lesiones cutáneas fueron reduciendo su tamaño e induración. A los 25 días de vida presentó hipercalcemia (valores de calcio de 13,48 mg/dL), pautándose tratamiento con sueroterapia al doble de las necesidades basales, furosemida y corticoterapia intravenosa, con lo que se normalizó la calcemia a las 24 horas. A pesar del tratamiento recibido, a las 72 horas apareció nuevamente un episodio de hipercalcemia, alcanzando cifras de calcio de 16,23 mg/dL, por lo que la paciente fue trasladada a la unidad pediátrica de cuidados intensivos para su monitorización. Ante la severidad del cuadro, se añadieron bisfosfonatos al tratamiento instaurado (de elección el pamidronato disódico), con lo que las cifras de calcio disminuyeron en 24 horas hasta valores normales. Se fueron reduciendo progresivamente las dosis de corticoides y furosemida. En los controles posteriores, las cifras de calcio en sangre se encontraban dentro del rango de la normalidad y la paciente permanecía asintomática, por lo que fue dada de alta con seguimiento en consultas externas, con control analítico semanal durante las primeras 6 semanas, y mensuales hasta 6 meses después de la resolución de las lesiones.
La NGS es una entidad infrecuente, relacionada con diferentes factores predisponentes5: maternos (diabetes gestacional, preeclampsia, uso de antagonistas del calcio, consumo de cocaína y tabaquismo...), complicaciones del parto (asfixia, aspiración de meconio, infecciones y traumatismo cutáneo) o factores neonatales6 (anemia, trombocitosis). Las lesiones típicas son placas eritematoso-violáceas con nódulos7, de consistencia dura y dolorosa. Debe hacerse el diagnóstico diferencial con distintas afecciones: escleredema neonatal, hemangioma, fibromatosis... A pesar de su carácter benigno y autorresolutivo, en ocasiones puede presentar complicaciones extracutáneas: trombocitopenia, hipoglucemia, hipertrigliceridemia e hipercalcemia (esta última infrecuente y de etiopatogenia desconocida), aunque la teoría más aceptada es la producción extrarrenal de 1,25 dihidroxivitamina D en los macrófagos del tejido granulomatoso. El tratamiento indicado ante una hipercalcemia8-10 es seguir una dieta baja en calcio, evitando la administración de vitamina D, y administrar hidratación intravenosa, acompañada de furosemida para forzar la calciuresis, y glucocorticoides, que interfieren en el metabolismo de la vitamina D y frenan la producción extrarrenal de 1,25 dihidroxivitamina D. En las hipercalcemias severas o persistentes puede ser útil administrar calcitonina y bisfosfonatos (de elección pamidronato). Debido a la gravedad de esta afección, aun encontrándose los pacientes asintomáticos, es necesario establecer una estrecha vigilancia hasta 6 meses después de la resolución de las lesiones.
Bibliografía
- Avayú H, Rodríguez C, Wortsman C, Corredoira S, Serman V, Strauch BG, et al. Necrosis grasa del recién nacido: a propósito de un caso. Rev Chil Pediatr. 2009; 80: 60-64.
- Grández N, Bravo F. Reporte de un caso de necrosis grasa subcutánea del recién nacido. Folia Dermatológica Peruana. 2004; 15: 2.
- Pardo R, Morán M, Álvarez CC, Solís G. Hipercalcemia mantenida secundaria a necrosis grasa subcutánea. Bol Pediatr. 2010; 50: 28-32.
- Karimi A, Sayyahfar S, Jadali F, Fahimzad A, Armin S, Ghorubi J, et al. Subcutaneous fat necrosis of the newborn complicated with hypercalcaemia. Pak J. Med Sci. 2008; 24: 178-180.
- Larralde M, Abad E, Corbella C, Ferrari CA, Plafnik R. Necrosis grasa subcutánea del recién nacido, comunicación de cinco casos. Dermatol Argent. 2009; 15: 200-204.
- Rivas AM, Vásquez LA, Molina V, Arredondo MI, Arroyave JE, Ruiz AC. Necrosis grasa del recién nacido asociada a anemia y trombocitopenia: reporte de un caso. Rev Asoc Col Dermatol. 2009; 17: 180-183.
- Tsuji T. Subcutaneous fat necrosis of the newborn: light and electron microscopio studies. Br J Dermatol. 1976; 95: 407-416.
- Alijaser M, Weinstein D. A 1-week-old newborn with hypercalcemia and palpable nodules: subcutaneous fat necrosis. CMAJ. 2008; 178: 1.653-1.654.
- Vijayakumar M, Prahlad N, Nammalwar BR, Shanmughasundharam R. Subcutaneous fat necrosis with hypercalcemia. Indian Pediatr. 2006; 43: 360-363.
- Dudink J, Walther FJ, Beekman RP. Subcutaneous fat necrosis of the newborn: hypercalcaemia with hepatic and atrial myocardial calcification. Arch Dis Child Fetal Neonatal. 2003; 88: 343F-345F.
Nefronía lobar aguda: a propósito de tres casos
La nefronía lobar aguda (NLA) es una infección bacteriana localizada en el parénquima renal, escasamente descrita en la literatura pediátrica. Presentamos tres casos de NLA, uno de ellos adquirido por vía hematógena. En los tres casos el diagnóstico se realizó mediante ecografía renal.
Queremos resaltar la utilidad de la ecografía renal para el diagnóstico precoz, así como la importancia de un adecuado tratamiento antibiótico.
Soporte nutricional en la enfermedad renal crónica
Los niños con una enfermedad renal crónica tienen un alto riesgo de desnutrición y requieren un soporte nutricional especializado, sobre todo en un estadio de la enfermedad ≥2. La pérdida de función de un órgano metabólicamente tan activo entraña alteraciones en el metabolismo intermediario de los nutrientes, así como en la biodisponibilidad y la pérdida de éstos. El riñón enfermo tiene una pérdida de función progresiva en la que están implicados muchos factores, entre los que el factor nutricional es importante.
El retraso de crecimiento es la afectación más importante durante la infancia. La alteración depende del grado de afectación y de la edad del paciente. El riesgo es mayor cuando la enfermedad es congénita, porque durante el primer año la velocidad de crecimiento es muy alta y los requerimientos nutricionales muy elevados y de difícil cobertura.
Las alteraciones motoras del tracto gastrointestinal producen anorexia y vómitos que dificultan la ingesta; por ello, estos pacientes frecuentemente requieren suplementación nutricional y una nutrición enteral prolongada mediante gastrostomía, que en general es endoscópica percutánea.
Administración de metamizol, insuficiencia renal y constricción intrauterina del conducto arterioso. Una asociación poco conocida
El metamizol, o dipirona magnésica, es un antiinflamatorio no esteroideo (AINE) ampliamente utilizado en Europa como analgésico y antipirético. Como con la indometacina y el ibuprofeno, se han descrito casos de oligoamnios, insuficiencia renal y cierre intrauterino del conducto arterioso fetal después del uso de metamizol en el tercer trimestre de la gestación, aunque su asociación con estos efectos adversos es menos conocida que con otros AINE. En el presente artículo presentamos un caso de hidropesía fetal con oligoamnios y constricción intrauterina ductal, que cursó con insuficiencia renal e hipertensión pulmonar, en el hijo de una madre que había recibido metamizol en el contexto de una pielonefritis aguda. Asimismo, se discutirá la fisiopatología de esta entidad y se revisará la bibliografía científica existente sobre el tema.
Micofenolato de mofetilo en cuatro pacientes con síndrome nefrótico e hialinosis segmentaria y focal
Describimos los casos de cuatro pacientes con síndrome nefrótico (SN) diagnosticados, mediante biopsia, de hialinosis segmentaria y focal (tres de ellos corticodependientes, con múltiples recaídas a pesar del tratamiento con corticoides e inmunosupresores, y uno de ellos corticorresistente), en los que se inició tratamiento con micofenolato de mofetilo (MMF). Durante el tratamiento se registraron los niveles plasmáticos de ácido micofenólico de forma mensual. Se controlaron en la consulta la función renal (mediante la fórmula de Schwartz) y la proteinuria, así como los posibles efectos secundarios de la medicación.
Nuestros pacientes presentaron sus primeros brotes de SN a edades similares (2-3 años). La edad de inicio del fármaco fue en dos pacientes antes de los 4 años y en los otros dos después de los 12 años. A pesar de usar dosis supraterapéuticas (mediana de los niveles de 5,625), todos los pacientes han mantenido una evolución clínico-analítica satisfactoria, con rangos de función renal y proteínas totales normales, sin presentar ninguna recaída (remisión total) o efectos secundarios (salvo gastrointestinales leves) ni precisar otras terapias asociadas. La duración media del tratamiento fue de 37,25 meses (rango: 16-56). Los hallazgos indican la eficacia y la seguridad de MMF en monoterapia en nuestros pacientes con SN e hialinosis segmentaria y focal en quienes han fracasado otros tratamientos.
Hematuria glomerular asociada a la gripe A H1N1
Sr. Director:
La gripe A, variedad H1N1, ha constituido una nueva epidemia que afectó de manera especial a niños y adolescentes, y de la que aún no conocemos en detalle sus diversas manifestaciones y sus posibles complicaciones1. Comunicamos, por su rareza, el caso de un varón de 12 años de edad que presentó un cuadro clínico de hematuria glomerular autolimitada no descrito o documentado hasta el momento.
El niño carecía de antecedentes patológicos y acudió a la consulta a finales de 2009 debido a una emisión súbita de orina colúrica, sin antecedentes de traumatismo renal. No presentaba fiebre, exantemas, dolor en la fosa renal ni síntomas urinarios en las vías bajas. Cinco días antes fue diagnosticado de amigdalitis y aún seguía tratamiento con amoxicilina-clavulánico. En la exploración se observaron los siguientes parámetros: peso de 66 kg (>p97), talla de 166 cm (>p97), índice de masa corporal de 24 kg/m2, perímetro craneal de 56 cm, presión arterial de 125/62 mmHg; el resto de la exploración general no presentaba datos relevantes. Los estudios complementarios mostraron los siguientes resultados: glucosa 100 mg/dL, BUN 29 mg/dL, creatinina 0,62 mg/dL, colesterol total 125 mg/dL, cLDL 63 mg/dL, cHDL 44 mg/dL, triglicéridos 88 mg/dL, calcio 9,26 mg/dL, fósforo 3,73 mg/dL, magnesio 2,45 mg/dL, proteínas totales 7,86 mg/dL, albúmina 4 g/dL, sodio 142 mEq/L, potasio 4,7 mEq/L, cloro 105 mEq/L, GOT 40 UI/L, GPT 46 UI/L, γ-GT 13 UI/L, FA 208 UI/L, PCR 1,1 mg/L, ASLO 280 UI/mL, factor reumatoide 8 UI/L, IgA 128 mg/dL, C3 123 mg/dL (90-180), C4 29 mg/dL (10-40), CH50 59 UI/mL (normal >24 UI/mL); velocidad de sedimentación globular 6 mm/h; ANA negativos; índice de Quick del 96%. En el test de orina al ingreso (tira reactiva) se detectó una densidad de 1.020, un pH de 5, 25 leucocitos/μL y 250 hematíes/ μL. En el sedimento de orina al ingreso se obtuvieron los siguientes parámetros: 2 leucocitos/campo, intensa hematuria (50% dismórficos), y a las 24 horas de evolución, 5 hematíes/ campo. En la determinación de orina de 24 horas (2.080 mL) se obtuvieron los siguientes resultados: GFR 122 mL/min/1,73 m2, FENA 1,01%, RTP 92%, proteinuria 4,5 mg/m2/h y calciuria 1,7 mg/kg/h. En el frotis faríngeo se detectó una flora mixta bacteriana. La inmunofluorescencia en secreciones orofaríngeas fue positiva para influenza A y H1N1, y negativa para influenza B. La fijación del complemento en suero dio positivo a 1/128 para influenza A, y negativo para influenza B y adenovirus. La determinación de anticuerpos heterófilos, así como la serología IgM para citomegalovirus, toxoplasma, virus de Epstein-Barr y herpes tipos 1 y 2, fueron negativas. El urocultivo también fue negativo. En la ecografía renal se observaban unos riñones de tamaño, situación y ecoestructura normales. El paciente evolucionó sin incidencias. La hematuria macroscópica desapareció en 48 horas, y la microhematuria se negativizó en una semana. El paciente no recibió tratamiento con oseltamivir.
La gripe es una infección vírica respiratoria frecuente en niños1. En una serie de 745 niños con gripe, las complicaciones más habituales fueron las siguientes: neumonía bacteriana (15%), convulsión (7%), insuficiencia respiratoria (4%), miositis (2%), encefalopatía (1%) y bacteriemia (1%)2-4. También se han descrito casos de hepatitis y encefalitis5.
El paciente presentó un cuadro de hematuria macroscópica con hematíes dismórficos, que se resolvió rápida y espontáneamente en el seno de una gripe A H1N1. Por ello, consideramos que esta última pudo ser el agente causal de una glomerulonefritis aguda, aunque leve. La glomerulonefritis suele manifestarse como un síndrome nefrítico agudo, de gravedad variable: la hematuria está presente en el 100% de los casos (macroscópica en el 75-90%), la proteinuria suele ser <500 mg/ dL, la función renal abarca desde la normalidad hasta el fallo renal grave, y la hipertensión arterial se presenta en el 60-80% de casos6,7. Podemos constatar la presencia de la gripe pandémica H1N1 por la coincidencia en el tiempo de inmunofluorescencia positiva para H1N1 en secreciones respiratorias y positividad para la gripe A en suero de la fase aguda. Desde el punto de vista epidemiológico, el paciente ingresó en la semana número 46 de 2009, que fue una de las semanas con una mayor tasa de incidencia de gripe y de la nueva gripe en España8 y en la Comunidad Valenciana, con una elevada incidencia en el grupo de niños de 5-14 años de edad9.
Por lo que se refiere a la asociación entre la gripe y la glomerulonefritis, una serie antigua comunica como complicación la insuficiencia renal aguda con oliguria y hematuria10. En un trabajo realizado en nueve soldados con una enfermedad respiratoria no estreptocócica y glomerulonefritis, algunos individuos presentaron datos serológicos de gripe A y B. Los autores concluyeron que las infecciones respiratorias de las vías altas no estreptocócicas frecuentemente se asociaban con glomerulonefritis, y las alteraciones histológicas y de la función renal pueden tardar unos 2-8 meses en normalizarse11.
Bibliografía
1. Cruz-Cañete M, Moreno-Pérez D, Jurado-Ortiz A, García-Martín F, López-Siles J, Olalla-Martín L. El virus de la gripe en pediatría. Un motivo de hospitalización. Enferm Infecc Microbiol Clin. 2007; 25: 177-183.
2. Coffin SE, Zaoutis TE, Wheeler Rosenquist AB, Heydon K, Herrera G, Bridges CB, et al. Incidence, complications, and risk factors for prolonged stay in children hospitalized with community-acquired influenza. Pediatrics. 2007; 119: 740-748.
3. Peltola V, Ziegler T, Ruuskanen O. Influenza A and B virus infection in children. Clin Infec Dis. 2003; 36: 299-305.
4. Dell KM, Schukman SL. Rhabdomyolisis and acute renal failure in a child with influenza A infection. Pediatr Nephrol. 1997; 11: 363- 365.
5. The epidemiology of influenza in children hospitalized in Canada, 2004-2005, in Immunization Monitoring Program Active (IMPACT) centres. CCDR. 2006; 32: 77-86.
6. Rodríguez-Iturbe B, Gordillo Paniagua G. Glomerulonefritis aguda. En: García Nieto V, Santos Rodríguez F, Rodríguez-Iturbe B, eds. Nefrología pediátrica, 2.ª ed. Madrid: Grupo Aula Médica, 2006.
7. Martín Govantes J. Glomerulonefritis aguda. Pediatr Integral. 2005; 9: 353-360.
8. Anónimo [internet]. Madrid: Ministerio de Sanidad y Política Social [citado el 12 de febrero de 2010]. Disponible en: http://www.msc. es/servCiudadanos/alertas/informesGripeA/home.htm
9. Anónimo [internet]. Generalitat Valenciana. Conselleria de Sanitat. Direcció General de Salut Pública. Àrea d'Epidemiologia [citado el 12 de febrero de 2010]. Disponible en: http://www.sp.san.gva.es/ DgspPortal/docs/semgripc.pdf
10. Jones SR. Potential complications of influenza A infections. Western J Med. 1976; 125: 341-346.
11. Smith MC, Cooke JH, Zimmerman DM, Bird JJ, Feaster BL, Morrison RE, et al. Asymptomatic glomerulonephritis after nonstreptococcal upper respiratory infections. Ann Intern Med. 1979; 91: 697-702.
Síndrome lumbocostovertebral
Sr. Director:
Presentamos el caso de una recién nacida sin exposición prenatal a teratógenos ni antecedentes familiares de interés, que presenta una tumoración blanda en el hipocondrio derecho que protruye con el llanto, y otra más pequeña en el quinto espacio intercostal derecho (figura 1). En el diagnóstico por la imagen destaca una herniación de la pared abdominal derecha de 10 mm, a través de la cual se hernia el lóbulo hepático al aumentar la presión abdominal, y una agenesia renal derecha. Radiográficamente se constata la ausencia del cuarto arco costal anterior y la presencia de anomalías vertebrales de la quinta a la décima vértebra torácica (figura 2). El cariotipo era 46,XX. Se realizó cirugía reparadora de la hernia de la pared abdominal a los 6 meses; la función renal, el desarrollo físico y psicomotor eran normales al año de edad.
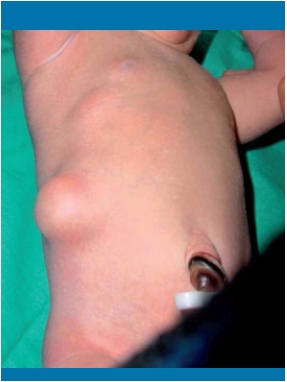
Figura 1. Tumoración blanda en el hipocondrio derecho que protruye con el llanto, y otra de menor tamaño en el quinto espacio intercostal derecho
|
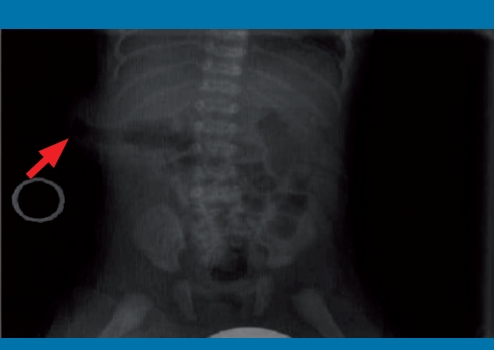
Figura 2. Herniación de la pared abdominal derecha de 10 mm, a través de la cual se hernia el lóbulo hepático al aumentar la presión abdominal
|
La hernia lumbar congénita es un defecto infrecuente de la pared que puede aparecer en cualquier localización comprendida entre la duodécima costilla y la cresta iliaca. Puede aparecer en el triángulo superior (hernia de Grynfelt Lesshaft), que es la más habitual, o en el inferior (triángulo de Petit)1. Clínicamente se manifiesta como la protrusión de una masa en el flanco que aumenta con el llanto y se reduce fácilmente1.
El síndrome lumbocostovertebral (SLCV) es muy poco frecuente y escasamente conocido (se han descrito unos 20 casos hasta la actualidad), y se caracteriza por la aparición de anomalías costovertebrales y de la musculatura del tronco. Incluye hemivértebras, ausencia de costillas, mielomeningocele anterior e hipoplasia de la pared anterior abdominal, que se manifiesta como una hernia lumbar congénita. También se han descrito casos de disrafismo espinal posterior, hepatopatía, artrogriposis, cardiopatía y agenesia renal como defectos asociados2-5.
Se postula que este síndrome puede deberse a un único defecto que ocurre entre la tercera y la quinta semanas de gestación, tiempo en que el mesodermo, situado entre el ectodermo y el endodermo a ambos lados de la notocorda, se diferencia en somitas, dando lugar al esclerotomo (procesos costales y vertebrales), el miotomo (musculatura esquelética del tronco) y el dermatomo (capas profundas de la piel y tejido celular subcutáneo)2. La disgenesia del primer miotomo lumbar sería responsable de la hernia lumbar en estos pacientes. Entre las semanas tercera y quinta se cierra el tubo neural y se produce la agenesia renal, defectos descritos en el SLCV.
Este tipo de anomalías se han descrito en hijos de madres diabéticas y en casos de exposición intrauterina al ácido lisérgico5. En animales, la exposición intrauterina en la rata a la purina 2-cloro-2-deoxiadenosina, un antimetabolito empleado en el tratamiento de algunas leucemias, reproduce en los fetos el espectro de las malformaciones observadas en el SLCV6.
Ante la presencia de una hernia lumbar congénita, que es un defecto poco habitual pero evidente clínicamente, además de descartar las posibles exposiciones intrauterinas, y dada la gran relación existente entre este defecto y el SLCV, es importante descartar la presencia de anomalías asociadas previamente a su reparación quirúrgica, que se aconseja realizar durante el primer año de vida.
1. Peláez Mata DJ, Álvarez Muñoz V, Fernández Jiménez I, García Crespo JM, Teixidor de Otto JL. Hernia lumbar congénita. Cir Pediatr. 1998; 11: 126-128.
2. Touloukian RJ. The lumbocostovertebral syndrome: a single somatic defect. Surgery. 1972; 71: 174-181.
3. Tavares de la Paz LA, Martínez-Ordaz JL. Hernia lumbar. Información de un caso y revisión de la literatura. Cir Ciruj. 2007; 75: 381-384.
4. Akcora B, Temiz A, Babayigit C. A different type of congenital lumbar hernia associated with the lumbocostovertebral syndrome. J Pediatr Surg. 2008; 43: e21-3.
5. Bhat RY, Greenough A, Rafferty GF, Patel S, Chandler C. Assessment of diaphragm function in lumbocostovertebral syndrome. Eur J Pediatr. 2004; 163: 694-695.
6. Lau C, Narotsky MG, Lui D, Best D, Setzer RW, Mann PC, et al. Exposure-disease continuum for 2-chloro-2-'deoxyadenosine (2- CdA), a prototype teratogen: induction of lumbar hernia in the rat and species comparison for the teratogenic responses. Teratology. 2002; 66: 6-18.
Tratamiento de las infecciones urinarias en pediatría con extracto de arándano rojo americano
Introducción: En la búsqueda de alternativas a la quimioprofilaxis antibiótica en la prevención de las infecciones urinarias (ITU) en el niño, hemos comenzado a utilizar un extracto de arándano rojo americano con 118 mg de proantocianidinas. Éstas inhiben la adherencia a la pared de la vía urinaria de Escherichia coli fimbriada tipo P.
Objetivos: Observar la eficacia y la tolerancia de un extracto de arándanos en niños con ITU frecuentes.
Material y métodos: Se seleccionan grupos de pacientes con ITU recidivantes frecuentes sin patología orgánica malformativa ni vejiga neuropática, litiasis o insuficiencia renal. El estudio observacional se efectuó durante 1 año en 62 niños de 5-17 años de edad.
Resultados: Se obtuvo un 100% de prevención de la pielonefritis aguda y un 92% de ausencia de infecciones sintomáticas.
Conclusiones: Hemos observado una gran eficacia del producto, ausencia de efectos adversos y una muy buena aceptación por parte de los padres y los pacientes para realizar un tratamiento prolongado, así como una baja tasa de abandonos.
Será necesario realizar estudios prospectivos, doble ciego, aleatorizados y controlados con placebo para poder establecer recomendaciones con un alto grado de evidencia.
Diagnóstico, tratamiento y prevención de la otitis media aguda en la infancia
La otitis media aguda (OMA) es el diagnóstico más frecuente en la consulta de pediatría de atención primaria y motivo habitual de prescripción de antibióticos. La prevalencia mundial de la otitis media es elevada, sobre todo durante la primera infancia. De cada cinco infecciones respiratorias de las vías altas aproximadamente una se complica con un episodio de OMA. El pico de incidencia se produce entre los 6 y los 12 meses. La mayoría de los niños padecen al menos un episodio antes de la edad escolar, y un tercio de ellos desarrolla una OMA recurrente. Aunque pocos episodios de OMA producen complicaciones graves, como mastoiditis y meningitis, muchos otros pueden provocar una disminución de la audición secundaria, debido a la persistencia de líquido en el oído medio. Esta hipoacusia, intermitente o crónica, puede dar lugar a trastornos del aprendizaje y a problemas en el desarrollo del lenguaje.








